Генетические нарушения у человека и методы их выявления

Генами называются участки ДНК, в которых закодирована структура всех белков в теле человека или любого другого живого организма. В биологии действует правило: «один ген – один белок», то есть в каждом гене содержится информация только об одном определенном белке.
В 1990 году большая группа ученых из разных стран начала проект под названием «Геном человека». Он завершился в 2003 году и помог установить, что человеческий геном содержит 20–25 тысяч генов. Каждый ген представлен двумя копиями, которые кодируют один и тот же белок, но могут немного различаться. Большинство генов одинаковые у всех людей – различается всего 1%.
ДНК находится в клетке внутри ядра. Она особым образом организована в виде хромосом – эти нитеподобные структуры можно рассмотреть в микроскоп с достаточно большим увеличением. Внутри хромосомы ДНК намотана на белки – гистоны. Когда гены неактивны, они расположены очень компактно, а во время считывания генетического материала молекула ДНК расплетается.
В клетках человека есть структуры, которые называются митохондриями. Они выполняют роль «электростанций» и отвечают за дыхание. Это единственные клеточные органеллы, у которых есть собственная ДНК. И в ней тоже могут возникать нарушения. 
Весь набор хромосом в клетке называется кариотипом. В норме у человека он представлен 23 парами хромосом, всего их 46. Выделяют два вида хромосом:
Методы исследования хромосом
Для исследования кариотипа применяют специальный метод – световую микроскопию дифференциально окрашенных метафазных хромосом культивированных лимфоцитов периферической крови.
Этот анализ применяется для диагностики различных хромосомных заболеваний. Он позволяет выявлять такие нарушения, как:

Однако с помощью исследования кариотипа можно выявить не все генетические нарушения. Оно не способно обнаружить такие изменения, как:

Для получения дополнительной информации, не видимой в световой микроскоп, используют хромосомный микроматричный анализ (ХМА). С его помощью можно изучить все клинически значимые участки генома и выявить изменения в количестве и структуре хромосом, а именно микрополомки (микроделеции и микродупликации).
Во время хромосомного микроматричного анализа применяют технологию полногеномной амплификации и гибридизации фрагментов опытной ДНК с олигонуклеотидами, нанесенными на микроматрицу. Если объяснять простыми словами, то сначала ДНК, которую необходимо изучить, копируют, чтобы увеличить ее количество, а затем смешивают ее со специальными ДНК-микрочипами, которые помогают выявлять различные нарушения.
Эта методика позволяет в одном исследовании выявлять делеции и дупликации участков ДНК по всему геному. Разрешающая способность стандартного ХМА от 100 000 пар нуклеотидов – «букв» генетического кода (в отдельных регионах от 10 000 п. н.).
С помощью ХМА можно выявлять:
Однако, как и предыдущий метод, хромосомный микроматричный анализ имеет некоторые ограничения. Он не позволяет выявлять или ограничен в выявлении таких аномалий, как:
Мутации в генах и заболевания, к которым они способны приводить
Мутации – это изменения, которые происходят в ДНК как случайным образом, так и под действием разных факторов, например химических веществ, ионизирующих излучений. Они могут затрагивать как отдельные «буквы» генетического кода, так и большие участки генома. Мутации происходят постоянно, и это основной двигатель эволюции. Чаще всего они бывают нейтральными, то есть ни на что не влияют, не приносят ни вреда, ни пользы. В редких случаях встречаются полезные мутации – они дают организму некоторые преимущества. Также встречаются вредные мутации – из-за них нарушается работа важных белков, наоборот, происходят достаточно часто. Генетические изменения, которые происходят более чем у 1% людей, называются полиморфизмами – это нормальная, естественная изменчивость ДНК Полиморфизмы ответственны за множество нормальных отличий между людьми, таких как цвет глаз, волос и группа крови.
Все внешние признаки и особенности работы организма, которые человек получает от родителей, передаются с помощью генов. Это важнейшее свойство всех живых организмов называется наследственностью. В зависимости от того, как проявляются гены в тех или иных признаках, их делят на две большие группы.
Например, карий цвет глаз у человека является доминантным. Поэтому у кареглазых родителей с высокой вероятностью родится кареглазый ребенок. Если у одного из родителей глаза карие, а у другого голубые, то вероятность рождения кареглазых детей в такой семье тоже высока. У двух голубоглазых родителей, скорее всего, все дети тоже будут голубоглазыми. А вот у кареглазых родителей может родиться ребенок с голубыми глазами, если у обоих есть рецессивные «гены голубоглазости», и они достанутся ребенку. Конечно, это упрощенная схема, потому что за цвет глаз отвечает не один, а несколько генов, но на практике эти законы наследования зачастую работают. Аналогичным образом потомству могут передаваться и наследственные заболевания. 
Как выявляют рецессивные мутации?
Для выявления мутаций, которые передаются рецессивно, используют целый ряд исследований.
Секвенирование по Сэнгеру – метод секвенирования (определения последовательности нуклеотидов, буквально – «прочтение» генетического кода) ДНК, также известен как метод обрыва цепи. Анализ используется для подтверждения выявленных мутаций. Это лучший метод для идентификации коротких тандемных повторов и секвенирования отдельных генов. Метод может обрабатывать только относительно короткие последовательности ДНК (до 300–1000 пар оснований) одновременно. Однако самым большим недостатком этого метода является большое количество времени, которое требуется для его проведения.
Если неизвестно, какую нужно выявить мутацию, то используют специальные панели.
Панель исследования — тестирование на наличие определенных мутаций, входящих в перечень конкретной панели исследования. Анализ позволяет выявить одномоментно разные мутации, которые могут приводить к генетическим заболеваниям. Анализ позволяет компоновать мутации в панели по частоте встречаемости (скрининговые панели, направленные на выявление носительства патологической мутации, часто встречаемой в данном регионе или в определенной замкнутой популяции) и по поражаемому органу или системе органов (панель «Патология соединительной ткани»). Но и у этого анализа есть ограничения. Анализ не позволяет выявить хромосомные аберрации, мозаицизм и мутации, не включенные в панель, митохондриальные заболевания, а также эпигенетические нарушения.
Не в каждой семье можно отследить все возможные рецессивные заболевания. Тогда на помощь приходит секвенирование экзома – тест для определения генетических повреждений (мутаций) в ДНК путем исследования в одном тесте практически всех областей генома, кодирующих белки, изменения которых являются причиной наследственных болезней.
Секвенирование следующего поколения-NGS – определение последовательности нуклеотидов в геномной ДНК или в совокупности информационных РНК (транскриптоме) путем амплификации (копирования) множества коротких участков генов. Это разнообразие генных фрагментов в итоге покрывает всю совокупность целевых генов или, при необходимости, весь геном.
Анализ позволяет выявить точечные мутации, вставки, делеции, инверсии и перестановки в экзоме. Анализ не позволяет выявить большие перестройки; мутации с изменением числа копий (CNV); мутации, вовлеченные в трехаллельное наследование; мутации митохондриального генома; эпигенетические эффекты; большие тринуклеотидные повторы; рецессивные мутации, связанные с Х-хромосомой, у женщин при заболеваниях, связанных с неравномерной Х-деактивацией, фенокопии и однородительские дисомии, и гены, имеющие близкие по структуре псевдогены, могут не распознаваться. 
Что делать, если в семье есть наследственное заболевание?
Существуют два способа выявить наследственные генетические мутации у эмбриона:
Предимплантационное генетическое тестирование (ПГТ) в цикле ЭКО. Это диагностика генетических заболеваний у эмбриона человека перед имплантацией в слизистую оболочку матки, то есть до начала беременности. Обычно для анализа проводится биопсия одного бластомера (клетки зародыша) у эмбриона на стадии дробления (4–10 бластомеров). Существует несколько видов ПГТ: на хромосомные отклонения, на моногенные заболевания и на структурные хромосомные перестройки. Данные Simon с соавторами (2018) говорят о том, что в случае проведения ЭКО с ПГТ у пациентки 38–40 лет результативность ЭКО составляет 60%. Но при исследовании эмбриона есть ряд ограничений. Так, из-за ограниченного числа клеток можно не определить мозаицизм.
Если нет возможности провести ЭКО с ПГТ, то используют второй вариант – исследование плодного материала во время беременности.
Для забора плодного материала используют инвазивные методы:
Далее эти клетки исследуют при помощи одного или нескольких генетических тестов (которые имеют свои ограничения). Проведение инвазивных методов может быть связано с риском для беременности порядка 1%.
Таким образом, проведя дополнительные исследования, можно значительно снизить риск рождения ребенка с генетическим заболеванием в конкретной семье. Но привести этот риск к нулю на сегодняшний день, к сожалению, невозможно, так как любой генетический тест имеет ряд ограничений, что делает невозможным исключить абсолютно все генетические болезни.

Автор статьи
Пелина Ангелина Георгиевна
Ведёт генетическое обследование доноров Репробанка, осуществляет подбор доноров для пар, имеющих ранее рождённых детей с установленной генетической патологией.
Сколько у нас генов?
Сколько у нас генов?
Найти ответ на этот вопрос оказалось куда сложнее, чем кто-либо предполагал
Автор
Редактор
Статья на конкурс «био/мол/текст»: Это интересный вопрос, ответ на который должен был дать проект «Геном человека», завершившийся в 2003 году. После того как ученые получили основную информацию о геноме человека, они попытались определить число генов, но эта задача оказалось не такой простой. Цель настоящей статьи — суммировать и проанализировать научные данные по составлению каталога генов у человека.

Конкурс «био/мол/текст»-2018
Эта работа опубликована в номинации «Свободная тема» конкурса «био/мол/текст»-2018.
Генеральный спонсор конкурса — компания «Диаэм»: крупнейший поставщик оборудования, реагентов и расходных материалов для биологических исследований и производств.
Спонсором приза зрительских симпатий выступил медико-генетический центр Genotek.
Как же мало известно о генах! Первый раз я остро ощутила это, находясь на практике в лаборатории медицинской генетики Харбинского медицинского университета. Исследовательская группа, где я проходила стажировку, занималась изучением онкогена Sei-1, который индуцирует образование двухминутных хромосом (DM), что способствует развитию онкогенеза. Однако механизм образования онкогена Sei-1 остается неизвестным до сих пор. А ведь различные мутации генов являются причиной возникновения и других опасных заболеваний человека, помимо рака. Итак, в данной статье мы изложим некоторые соображения о том, почему мы все еще многое не знаем о генах, а также сформулируем наше мнение о том, сколько генов у человека.
В 1977 году Фредерик Сэнгер впервые разработал метод секвенирования ДНК [1], основанный на терминации ДНК-полимеразной реакции с помощью дидезоксинуклеотидов, за что в 1980 году был удостоен Нобелевской премии в области химии. В этом же году Нобелевскую премию получили Максам и Гилберт, которые предложили метод секвенирования ДНК путем химической деградации. В 1985 году была выявлена первая полная последовательность ДНК бактерии (Haemophilus influenzae), в 1996 году получен первый геном эукариотической клетки (дрожжи Saccharomyces ceravisiae), а в 1998 году расшифрован геном дождевого червя (Caenorhabditis elegans). Завершение в 2003 году проекта «Геном человека» привело к публикации полной последовательности человеческого генома. Но «полной» ее можно назвать весьма условно, учитывая, что около 8% не секвенировано и по сей день [2].
Проект «Геном человека» и полный список генов
Выявление полного списка генов необходимо для выяснения молекулярных механизмов возникновения и развития рака, шизофрении [3], деменции, а также многих других заболеваний человека. Секвенирование ДНК, выделенной из тканей больных, позволяет выявлять такие мутации, как нуклеотидные замены, делеции и вставки, ответственные за возникновение этих заболеваний.

Рисунок 1. Арт-проект на выставке «Геном — расшифровка кода жизни» в Национальном музее естественной истории в Вашингтоне
Собственно, ради этого и затевался проект «Геном человека» (Human genome project, HGP), который продолжался с 1990 по 2003 год. Его основной задачей было определение нуклеотидной последовательности ДНК человека и локализации 100 000 человеческих генов (как тогда полагали) [4]. Параллельно с этим планировалось изучить ДНК набора модельных организмов, чтобы получить сравнительную информацию, необходимую для понимания функционирования генома человека. Предполагалось, что информация, полученная в результате HGP, станет настольной книгой для биомедицинской науки в XXI веке [5]. Целями данных исследований являлось получение информации о причинах ряда болезней [6] и, в конечном итоге, разработка способов лечения более чем 4000 генетических заболеваний, которые затрагивают человечество, включая многофакторные, в которых генетическая предрасположенность играет важную роль. Считалось, что результаты секвенирования генома позволят определить локализацию каждого гена и их общее количество. Однако последовавшие за этим события доказали обратное: сегодня существует несколько баз данных генов, которые существенно отличаются друг от друга. Причем число белок-кодирующих генов совпадает, а число генов других типов расходится.
Проект «Протеом человека»
В 2010 году по инициативе Организации по изучению протеома человека (Human proteome organization, HUPO) был начат проект «Протеом человека» (HPP), целью которого является создание полного списка белков вида Homo sapiens [7]. Для этого, во-первых, предполагается идентифицировать и охарактеризовать, по крайней мере, по одному белковому продукту белок-кодирующих генов, их однонуклеотидные полиморфизмы и варианты сплайсинга, а также виды посттрансляционной модификации белков [8]. Во-вторых, данные протеомики, полученные в результате реализации HPP, способствуют, в дополнение к геномным данным, решению различных биомедицинских задач и созданию новых аннотированных баз знаний, таких как neXtProt [9].
В настоящее время neXtProt содержит информацию о 17 487 белках, существование которых экспериментально подтверждено, 1728 белках, подтвержденных на уровне транскриптов, 515-и, определенных на основании гомологии, 76-и предсказанных и 571-м неизвестной природы. Особый интерес вызывают белки, существование которых экспериментально не доказано, хотя данные о том, что они кодируются геномом, существуют. Это так называемые «потерянные» белки, которые составляют примерно 18% всех кодируемых белков. Для выявления и характеристики таких белков создан ресурс MissingProteinPedia [7].
«Протеом человека» является продолжением проекта «Геном человека». Предполагается, что благодаря проекту по изучению протеома мы узнаем точное количество белок-кодирующих генов, что впоследствии позволит понять, сколько всего генов у человека.
Немного о РНК
Проект «Геном человека» показал, что молекулы РНК также важны для жизни, как и ДНК. Внутри клеток существует множество РНК (рис. 2). Изначально РНК подразделяются на некодирующие РНК (нкРНК), которые не транслируются в белки, и кодирующие РНК (мРНК), служащие матрицей для синтеза полипептидных цепей белка. Некодирующие РНК имеют более сложную классификацию. Они бывают инфраструктурными и регуляторными. Инфраструктурные РНК представлены рибосомными РНК (рРНК) и транспортными РНК (тРНК). Молекулы рРНК синтезируются в ядрышке и составляют основу рибосомы, а также кодируют белки субъединиц рибосомы. После того, как рРНК полностью собраны, они переходят в цитоплазму, где в качестве ключевых регуляторов трансляции, участвуют в чтении кода мРНК. Последовательность из трех азотистых оснований в мРНК указывает на включение определенной аминокислоты в последовательность белка. Молекулы тРНК, приносят указанные аминокислоты на рибосомы, где синтезируется белок.
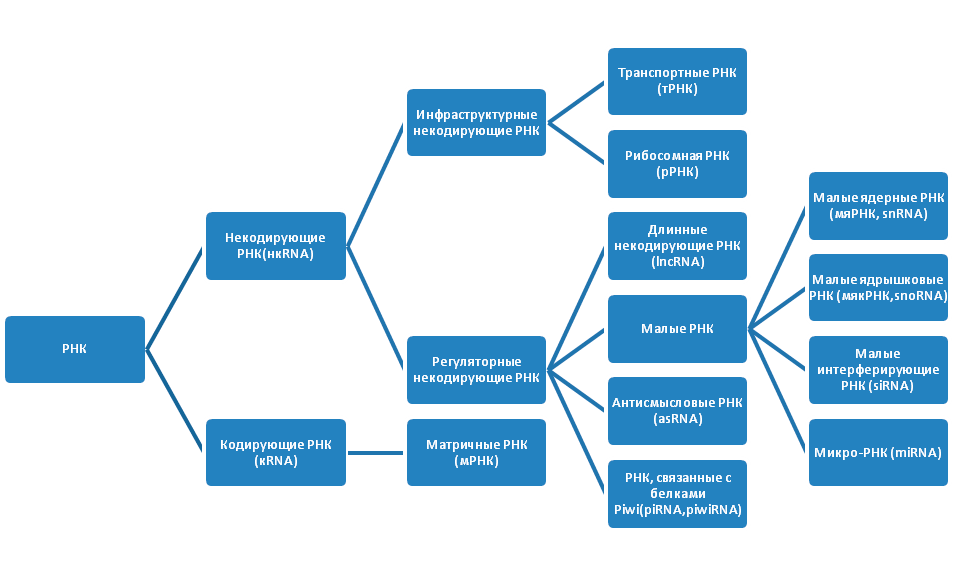
Рисунок 2. Виды РНК
рисунок автора статьи
Регуляторные нкРНК очень широко представлены в организме, классифицируются в зависимости от размера и выполняют ряд важных функций (табл. 1).
| Название | Обозначение | Длина | Функции | |
|---|---|---|---|---|
| Длинные некодирующие РНК | днкРНК, lncRNA | 200 нуклеотидов | 1. Регулируют избирательное метилирование ДНК, направляя ДНК-метилтрансферазу 2. Руководят избирательной посадкой репрессорных комплексов polycomb | |
| Малые РНК | Малые ядерные РНК | мяРНК, snRNA | 150 нуклеотидов | 1. Участвуют в сплайсинге 2. Регулируют активность факторов транскрипции 3. Поддерживают целостность теломер [13] |
| Малые ядрышковые РНК | мякРНК, snoRNA | 60–300 нуклеотидов | 1. Участвуют в химической модификации рРНК, тРНК и мяРНК 2. Возможно, участвуют в стабилизации структуры рРНК и защите от действия гидролаз | |
| Малые интерферирующие РНК | миРНК, siRNA | 21–22 нуклеотидов | 1. Осуществляют антивирусную иммунную защиту 2. Подавляют активность собственных генов | |
| Микро-РНК | мкРНК, miRNA | 18–25 нуклеотидов | Подавляют трансляцию путем РНК-интерференции | |
| Антисмысловые РНК [14] | asRNA | 1. Короткие: менее 200 нуклеотидов 2. Длинные: более 200 нуклеотидов | Блокируют трансляцию, образуя гибриды с мРНК | |
| РНК, связанные с белками Piwi | piRNA, piwiRNA | 26–32 нуклеотидов | Их также называют «стражами генома», они подавляют активность мобильных генетических элементов во время эмбриогенеза | |
Проблема терминологии
Прежде чем ответить на вопрос: «Сколько у нас генов?», нужно понять, что же такое ген?
Основное внимание HGP было направлено на белок-кодирующие гены [15]. Однако, как было указано в первоначальном докладе HGP в 2001 году, «тысячи генов человека продуцируют некодирующие РНК (нкРНК), являющиеся их конечным продуктом», хотя на тот момент было известно около 706 генов нкРНК [2]. В своей недавней статье, опубликованной в журнале BMC Biology Стивен Зальцберг (Steven L. Salzberg) дает следующее определение гена [16]:
Ген любой участок хромосомной ДНК, который транскрибируется в функциональную молекулу РНК или сначала транскрибируется в РНК, а затем транслируется в функциональный белок.
Это определение включает как гены некодирующих РНК, так и белок-кодирующие гены, и позволяет определять все варианты альтернативного сплайсинга в одном локусе как варианты одного и того же гена. Это позволяет исключить псевдогены – нефункциональные остатки структурных генов, утратившие способность кодировать белок.
Результаты двух первых исследований свидетельствовали о наличии у человека 31 000 [2] и 26 588 белок-кодирующих генов [17], а в 2004 появилась полная последовательность генома человека [4], и авторы подсчитали, что полный каталог насчитывает 24 000 белок-кодирующих генов. Каталог человеческих генов Ensembl включает 22 287 белок-кодирующих генов и 34 214 транскриптов [18].
Секвенирование нового поколения (NGS)
Появление высокопроизводительных методов параллельного секвенирования (в ходе такого секвенирования миллионы фрагментов ДНК из одного образца секвенируются одновременно) или секвенирования нового (следующего) поколения (next-generation sequencing, NGS) [1] позволило значительно ускорить поиск функциональных участков генома [4]. Биотехнологические компании разработали и коммерциализировали различные платформы для NG-секвенирования, позволяющие секвенировать от 1 млн до десятков млрд коротких последовательностей (ридов, reads) длиной 50–600 нуклеотидов каждая. К наиболее популярным платформам относятся такие, как Illumina и IonTorrent, использующие амплификацию ДНК с помощью ПЦР [19], а также платформы одномолекулярного секвенирования, такие как Helicos Biosciences HeliScope, Pacific Biosciences SMRT (single molecule real-time sequencing), и нанопорового секвенирования Oxford Nanopore, осуществляющие секвенирование в реальном времени и позволяющие прочитывать значительно более длинные риды — до 10–60 тыс. нуклеотидов. Кроме того, изобретение секвенирования РНК (RNA-seq) в 2008 году, которое создавалось для количественного определения экспрессии генов, также способствовало обнаружению транскрибируемых последовательностей, как кодирующих, так и некодирующих РНК [20].
Благодаря NGS, базы данных днкРНК и других генов РНК (таких как микро-РНК) резко выросли за десятилетие, и текущие каталоги генов человека теперь содержат больше генов, кодирующих РНК, чем белки (табл. 2).
| Типы генов | Gencode | Ensembl | RefSeq | CHESS |
|---|---|---|---|---|
| Белок-кодирующие гены | 19 901 | 20 376 | 20 345 | 21 306 |
| Гены длинных некодирующих РНК | 15 779 | 14 720 | 17 712 | 18 484 |
| Антисмысловые РНК | 5501 | — | 28 | 2694 |
| Другие некодирующие РНК | 2213 | 2222 | 13 899 | 4347 |
| Псевдогены | 14 723 | 1740 | 15 952 | — |
| Общее число транскриптов | 203 835 | 203 903 | 154 484 | 323 827 |

Рисунок 3. Последовательность ДНК, получаемая после секвенирования человеческого генома
В ходе секвенирования РНК обнаружилось, что альтернативный сплайсинг, альтернативное инициирование транскрипции и альтернативное прерывание транскрипции проиcходят гораздо чаще, чем полагали, затрагивая до 95% человеческих генов. Следовательно, даже если известно местоположение всех генов, сначала нужно выявить все изоформы этих генов, а также определить, выполняют ли эти изоформы какие-либо функции или они просто представляют собой ошибки сплайсинга.
Базы данных генов человека
Задача по составлению каталога всех генов по-прежнему не решена. Проблема заключается в том, что за последние 15 лет только две исследовательские группы составили список доминантных генов: RefSeq, которая поддерживается Национальным центром биотехнологической информации (NCBI) при Национальных институтах здоровья (NIH), и Ensembl/Gencode, которая поддерживается Европейской молекулярно-биологической лабораторией (EMBL). Однако, несмотря на большой прогресс, сейчас в каталогах различается количество белок-колирующих генов, генов длинных некодирующих РНК, псевдогенов, а также варьирует количество антисмысловых РНК и других некодирующих РНК (табл. 2). Каталоги еще дорабатываются: например, в прошлом году сотни генов, кодирующих белок, были добавлены или удалены из списка Gencode. Эти разногласия объясняют проблему создания полного каталога человеческих генов.
В 2017 году была создана новая база данных генов человека — CHESS. Примечательно, что она включает все белок-кодирующие гены как Gencode, так и RefSeq, так что пользователям CHESS не нужно решать, какую базу данных они предпочитают. Бóльшее количество генов может вызывать больше ошибок, но создатели считают, что бóльший набор окажется полезным при исследовании болезней человека, которые еще не отнесены к генетическим. Набор генов CHESS в настоящее время в версии 2.0 еще не окончательный, и, безусловно, создатели работают над его усовершенствованием.
Таким образом, все еще неизвестно, сколько всего генов у человека. Существует ряд проблем, затрудняющих эту задачу. Например, многие гены (особенно, гены днкРНК), видимо, имеют высокую тканеспецифичность. Из этого следует, что пока ученые подробно не исследуют все типы клеток человека, они не могут быть уверены, что обнаружили все человеческие гены и транскрипты. Безусловно, сегодня знания о человеческих генах стали значительно обширнее, чем в начале проекта «Геном человека», а технологии совершеннее. Это дает надежду на то, что в скором времени мы узнаем точный ответ на поставленный вопрос.