Врожденная дисфункция коры надпочечников
Адреногенитальный синдром Hадпочечники секретируют кортикостероиды, которые по биологическим свойствам можно разделить на 4 группы: глюкокортикоиды, минералокортикоиды, андрогены и эстрогены. Синтез кортизола в надпочечниках плода под вли
 |
| Адреногенитальный синдром |
Hадпочечники секретируют кортикостероиды, которые по биологическим свойствам можно разделить на 4 группы: глюкокортикоиды, минералокортикоиды, андрогены и эстрогены.
Синтез кортизола в надпочечниках плода под влиянием адренокортикотропного гормона (АКТГ) начинается рано, в связи с этим дефект ферментных систем приводит к вирилизации до рождения ребенка. Так как к этому сроку формирование внутренних половых органов заканчивается, вирилизация затрагивает наружные половые органы.
Из различных ферментных дефектов недостаточность 21-гидроксилазы встречается наиболее часто и протекает в виде двух клинических форм: вирильного и сольтеряющего синдромов.
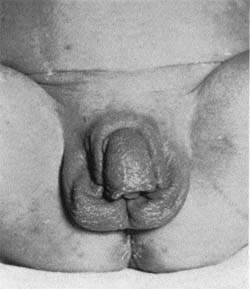
Вирильная форма связана с частичной недостаточностью 21-гидроксилазы. Как правило, при этом наблюдается компенсация функции коры надпочечников в результате повышенной секреции АКТГ, то есть уровень кортизола определяется на нижней границе нормы. Компенсаторное выделение АКТГ приводит к образованию значительного количества андрогенов, прогестерона, 17-гидроксипрогестерона, которые угнетают сользадерживающую активность на уровне канальцев почек, что ведет к значительному увеличению продукции альдостерона. У девочек с вирильной формой с рождения имеются признаки ложного гермафродитизма. Клитор гипертрофирован, напоминает по строению половой член, имеющий единое мочеполовое отверстие — урогенитальный синус, открывающийся у корня клитора, мошонкообразные большие половые губы.
| Врожденная дисфункция коры надпочечников является наследственным заболеванием, характеризующимся нарушением синтеза кортикостероидов, в основном кортизола, в результате дефицита ферментных систем коры надпочечников с одновременной избыточной продукцией андрогенов в надпочечниках |
У мальчиков с рождения отмечается увеличение полового члена, кожа мошонки морщинистая, выражена пигментация шва полового члена, кожи мошонки, передней линии живота, ареолы сосков.
В постнатальном периоде продолжается избыточная секреция андрогенов, усиливаются явления вирилизации. Дети быстрее растут, имеют развитую мускулатуру. Вначале больные обгоняют в росте сверстников, в дальнейшем же, после 9-10 лет, в связи с закрытием зон роста остаются низкорослыми. В возрасте 2–4 лет появляется преждевременное оволосение на лобке, подмышках, туловище, лице. У некоторых мальчиков значительно увеличивается половой член и появляются эрекции. Телосложение девочек мускулинного типа. В пубертатный период менструации не наступают, молочные железы не развиваются, так как повышенная секреция андрогенов надпочечниками по принципу обратной связи тормозит образование и выделение гонадотропинов. У мальчиков по этой причине угнетается развитие яичек — они остаются маленькими.
При сольтеряющей форме дефицит 21-гидроксилазы более глубокий, поэтому наряду с дефицитом образования кортизола происходит резкое снижение биосинтеза альдостерона. Если при вирильной форме потеря натрия, обусловленная избыточной продукцией прогестерона, 17-гидроксипрогестерона, компенсируется избыточной секрецией альдостерона, то при сольтеряющей форме снижено образование альдостерона. Результатом такого комбинированного действия является развитие клинической картины по типу острой надпочечниковой недостаточности.
| Заболевание наследуется по аутосомно-рецессивному типу и проявляется в гомозиготном состоянии. Распространенность составляет в среднем 1:4000-5000, а гетерозиготное носительство — 1:35-40 человек |
У новорожденных при этом синдроме резко выражены симптомы вирилизации, особенно у детей женского пола — полное заращение половой щели и появление мошоночноподобного образования. На 5–10-й день наступает быстрое ухудшение состояния, связанное с частой рвотой, жидким стулом. Развивается дегидратация, ребенок становится вялым, снижается масса тела. В сыворотке крови нарастает уровень кальция и снижается содержание натрия и хлора.
Гипертоническая форма дисфункции коры надпочечников, связанная с дефицитом 11b-гидроксилазы, встречается реже. В клинической картине, помимо признаков андрогенизации в раннем возрасте, характерно повышение артериального давления. При этой патологии отсутствует гиперплазия юкстагломерулярного аппарата почки и уровень ренина в плазме не повышен.
Дифференциальный диагноз врожденной дисфункции коры надпочечников у девочек первых лет жизни следует проводить с дисгенезией гонад, анорхизмом, истинным гермафродитизмом; у мальчиков — с преждевременным половым развитием различного генеза, в том числе с опухолью яичка, с андрогенпродуцирующей опухолью надпочечника (андростерома). Сольтеряющую форму у новорожденных дифференцируют с пилороспазмом и пилоростенозом.
У девочек старшего возраста при подозрении на врожденную дисфункцию коры надпочечников требуется исключить такие заболевания, как андрогенпродуцирующая опухоль яичника, стромальный текоматоз, поликистоз яичников.
Наиболее приемлемым средством заместительной терапии является преднизолон, который в течение длительного срока сохраняет достаточную концентрацию и обладает некоторым минералокортикоидным действием. Суточная доза его подбирается индивидуально и колеблется в зависимости от возраста: от 1,5–3 мг (для детей младше 3 лет) до 5 мг для детей старшего возраста. Реже применяют преднизолон, кортизон, дексаметазон. Соответственно суточному физиологическому ритму 17-гидроксипрогестерона принимать преднизолон рекомендуется рано утром, в 6-7 часов (причем в это время следует принять 2/3–1/2 дозы) и поздно вечером (23–24 часа). Глюкокортикоиды больные должны получать постоянно, так как отмена препарата ведет к декомпенсации заболевания.
При лечении больных с сольтеряющей формой врожденной дисфункции коры надпочечников к кортикостероидам добавляют минералкортикоиды. Это либо масляный раствор дезоксикортикостерона ацетата (ДОКА), либо таблетированный препарат кортинеф (0,1 мг), который в 5–10 раз активнее первого. Препараты назначаются в течение 1–2 недель ежедневно (ДОКА по 1–2 мл, кортинеф по 1 таб.) с последующим постепенным снижением дозы до оптимальной. Возможно поддерживающую терапию проводить через каждые 1–2 дня. У некоторых больных дефицит минералкортикоидов можно компенсировать добавлением в пищу поваренной соли. Обязательный контроль за весом, артериальным давлением, калием, натрием, ЭКГ.
Начальные дозы кортикостероидов должны быть в два раза выше физиологических.
При адекватном лечении у детей уменьшается мышечная гипертрофия, перераспределяется подкожножировая клетчатка, половое оволосение не прогрессирует. Если лечение начинают до закрытия ядер окостенения, то увеличивается темп роста. В случаях же поздней терапии эти дети так и остаются низкорослыми.
Гипертоническая форма заболевания лечится аналогично вирильной.
При присоединении интеркуррентных заболеваний дозу глюкокортикоидов увеличивают в 1,5–2 раза.
В случае развития острого криза недостаточности коры надпочечников показано внутривенное капельное введение жидкости (физиологический раствор и 5%-ный раствор глюкозы 100-200 мл/кг массы тела в сутки) в 4 приема, ДОКА 2–3 мг/кг массы тела в сутки. В последующие дни доза глюкокортикоидов быстро снижается и больной переводится на пероральный прием преднизолона.
Важное место в лечении врожденной дисфункции коры надпочечников занимает психотерапия, особенно в случае необходимости перемены пола ребенка.
Большинство девочек с этим заболеванием нуждается в проведении феминизирующей пластики наружных гениталий, удалении клитора и формировании малых половых губ после вскрытия урогенитального синуса. Коррекцию наружных гениталий рекомендуется проводить через год после назначения кортикостероидной терапии.
Адреногенитальный синдром: симптомы, лечение
Содержание
Какая связь между недостатком в организме гормона, контролирующего обмен углеводов и опосредующего стресс-реакции, и множественными нарушениями полового развития? Самая прямая. Примером служит т.н. адреногенитальный синдром у детей и взрослых. Этот довольно редкий наследственный недуг поражает людей обоего пола (в среднем — одного на пять–шесть тысяч) и уже в юном возрасте имеет свои характерные проявления.
Чем характеризуется? Кора надпочечников — важных «игроков» на «сцене» обмена веществ, процессов роста, развития, приспособления к меняющимся условиям и др., не поставляет организму должное количество химических регуляторов и координаторов этих процессов — кортизолаи, в некоторых случаях, альдостерона. Почему? Потому что в наличии врожденное нарушение образование необходимых для синтеза этих гормонов ферментов.
В результате, желая скомпенсировать эти недостатки, организм запускает каскад взаимодействий между различными органами, имеющий патологические следствия. Если более конкретно:
Формы врожденного адреногенитального синдрома определяются тем, какого (и в какой степени) фермента (или ферментов) организму не хватает. И, соответственно, какой конкретно гормон (или гормоны) находятся в дефиците. Чаще всего диагностируется т.н. вирильная форма. В самом названии (переводится как «мужская») кроется и комплекс сопутствующих ей симптомов — наличие вторичных мужских половых признаков у девочек и преждевременных гипертрофированных атрибутов сильного пола у мальчиков. В остальном недуг этой формы протекает довольно легко (т.н. компенсированная форма), поскольку работа фермента нарушена лишь частично, и при своевременном обнаружении хорошо поддается корректировке.
Две другие формы (редкие) — гипертоническая и сольтеряющая — более тяжелые. Настолько, что без их раннего выявления существует высокий риск гибели детей от сердечной и почечной недостаточности, и кровоизлияния в мозг.
Симптомы адреногенитального синдрома
Адреногенитальный синдром у новорожденных
Поскольку надпочечники под руководством мозговых центров (в ответ на нехватку кортизола) начинают усиленно трудиться еще внутриутробно, ребенок рождается на свет, уже имея избыток половых гормонов. В результате у новорожденных девочек регистрируют симптомы т.н. ложного гермафродитизма (наличия признаков обоих полов). А у новорожденных мальчиков — увеличенный копулятивный орган. Кроме того, аномальное количество половых стероидов приводит к избыточной пигментации наружных половых органов, кожи вокруг ануса и сосков. Перечисленные признаки характерны для самой распространенной (вирильной) формы адреногенитального синдрома.
При более редкой и тяжелой форме (сольтеряющей) к ним добавляются следствия потери организмом натрия и хлора, и задержки калия (из-за недостатка альдостерона):
Адреногенитальный синдром у девочек и мальчиков
С возрастом вышеуказанные признаки сохраняются (если не поставлен ранний диагноз и не начата заместительная терапия). К двум-четырем годам появляются симптомы преждевременного полового созревания с признаками маскулинизации (от латинского слова «мужчина, «самец») у девочек:
Касательно последнего пункта: высокие темпы роста характерны только для юного возраста. Довольно рано (к двенадцати годам) зоны роста закрываются, рост сильно притормаживается или останавливается вовсе. В результате в подростковом периоде наблюдается уже отставание в росте от сверстников.
Другими (неспецифическими) признаками не выявленных в очень раннем детстве гипертонической и сольтеряющей форм адреногенитального синдрома могут стать:
Длительная гипертензия рискует осложниться ухудшением зрения, сердечно-сосудистыми, почечными и мозговыми нарушениями (вплоть до инсульта).
Для правильной (и ранней) диагностики помимо осмотра и опроса необходимы лабораторные и инструментальные исследования:
Лечение адреногенитального синдрома
Чем раньше поставлен правильный диагноз, тем эффективнее лечение.
Прежде всего, оно состоит в заместительной терапии недостающими гормонами (на протяжении всей жизни человека). Хорошим подспорьем медикаментозным методам служат: диета с учетом формы патологии и индивидуальными особенностями организма и психотерапевтические сеансы. Возможно хирургическое вмешательство — удаление клитора, пластика влагалища.
Адреногенитальный синдром

Адреногенитальный синдром (АГС) также известен как врожденная дисфункция коры надпочечников (ВДКН) или врожденная гиперплазия коры надпочечников (ВГКН). Синдром объединяет заболевания с аутосомно-рецессивным типом наследования, в основе которых лежит дефект одного из ферментов метаболического пути синтеза стероидов.
Заболевание вызвано наследственным дефицитом ферментов, которые локализованы в коре надпочечников. Гормоны коры надпочечников включают минералокортикоиды (альдостерон), глюкокортикоиды (кортизол) и половые стероиды (тестостерон и эстроген). Синдром возникает, когда дефицит фермента приводит к снижению надпочечникового синтеза глюкокортикоидов. Происходит снижение ингибирующего влияния гормонов коры надпочечников на гипофиз, поэтому синтез и секреция адренокортикотропного гормона (АКТГ) увеличивается по механизму отрицательной обратной связи. АКТГ стимулирует увеличение размеров надпочечников и выработку ими промежуточных субстратов. Включаясь в другие пути синтеза, метаболиты вызывают повышение уровней других гормонов коры надпочечников — минералокортикоидов или андрогенов. Измененные уровни минералокортикоидов и половых гормонов приводят к аномалиям электролитного соотношения, проблемам с дифференциацией пола и другим признакам, и симптомам, в зависимости от дефицита фермента и его степени.
Частота встречаемости заболевания от 1:5000 до 1:67000.
Характеристика заболевания
ВГКН является одним из самых распространенных наследственных моногенных заболеваний, одновременно представляет собой вариант хронической первичной надпочечниковой недостаточности и группу патологии полового развития, а также половой дифференцировки. АГС включен в программу «Национальные приоритетные проекты» и введен в неонатальный скрининг. АГС в стертой (неклассической) форме является одной из причин нарушения репродуктивного здоровья (бесплодие, невынашивание беременности).
Патогенетической сущностью синдрома является нарушение процесса перехода 17-гидроксипрогестерона и 11-дезоксикортикостерона в 11-дезоксикортизол (рис 1). Развивается гормональная дисфункция, которая сводится к снижению синтеза кортизола альдостерона и избыточному накоплению предшественников кортизола. Избыток прегненолона, прогестерона, 17-гидроксипрогестерона конвертируется в надпочечениковые андрогены.
Рис.1. Недостаточность 21-гидроксилазы. Штриховкой выделены стероиды, синтез которых заблокирован.
 |
Так происходит угнетение выработки одних кортикостероидов при одновременном увеличении выработки других, вследствие дефицита того или иного фермента, обеспечивающего один из этапов стероидогенеза.
Симптомы недостаточности 21-гидроксилазы
При классической вирильной форме адреногенитального синдрома, наружные половые органы у девочек сформированы по гетеросексуальному типу — гипертрофирован клитор, большие половые губы напоминают мошонку, вагина и уретра представлены урогенитальным синусом. У новорождённых мальчиков явных нарушений выявить не удаётся. С 2–4 лет у детей обоего пола появляются другие симптомы адреногенитального синдрома, то есть андрогенизации: формируется подмышечное и лобковое оволосение, развивается скелетная мускулатура, грубеет голос, маскулинизируется фигура, появляются юношеские угри на лице и туловище. В пубертатном периоде у девочек не растут молочные железы, не появляются менструации. При этом ускоряется дифференцировка скелета, и зоны роста закрываются преждевременно, это обусловливает низкорослость.
При сольтеряющей форме 21-гидроксилазной недостаточности, помимо вышеописанных симптомов, у детей с первых дней жизни отмечают признаки надпочечниковой недостаточности. Появляются вначале срыгивания, затем рвоты, возможен жидкий стул. Ребёнок быстро теряет массу тела, развиваются симптомы дегидратации, нарушения микроциркуляции, снижается артериальное давление, начинается тахикардия, возможна остановка сердца вследствие гиперкалиемии.
Неклассическая форма адреногенитального синдрома характеризуется ранним появлением вторичного оволосения, ускорением роста и дифференцировки скелета. У девочек пубертатного возраста возможны умеренные признаки гирсутизма, нарушения менструального цикла (первый цикл может запаздывать или начаться раньше времени). В позднем возрасте при небольших дефектах гена на адреногенитальный синдром может указывать наличие бесплодия в анамнезе. Менструальный цикл может быть нерегулярным и иметь тенденцию к задержке. Невынашивание беременности при адреногенитальном синдроме встречается у четверти женщин с дефектным геном.
Публикации в СМИ
Синдром адреногенитальный
Адреногенитальный синдром — врождённое патологическое состояние, обусловленное дисфункцией коры надпочечников с чрезмерной секрецией андрогенов и проявляющееся признаками вирилизации.
Этиология и патогенез. Синдром обусловлен недостаточностью одного из ферментов, необходимых для синтеза кортизола. Дефицит кортизола стимулирует выработку АКТГ, что приводит к гиперплазии коры надпочечников и избыточной продукции АКТГ-зависимых стероидов, синтез которых при данной недостаточности фермента не нарушен (в основном, надпочечниковых андрогенов — дегидроэпиандростерона, андростендиона и тестостерона).
Генетические аспекты
• Недостаточность 21-гидроксилазы (*201910, КФ 1.14.99.10, 6p21.3, мутации генов CYP21, CA2, CYP21P, r ) наблюдают в 95% случаев адреногенитального синдрома •• При умеренном дефиците фермента развивается вирильная (неосложнённая) форма синдрома, при которой придают значение исключительно симптомам избытка андрогенов (глюкокортикоидная и минералокортикоидная недостаточность не проявляется) •• При полном блоке фермента развивается сольтеряющая (тяжёлая) форма (синдром Дебре–Фибигера). Наряду с уменьшением синтеза кортизола снижена продукция альдостерона; дефицит минералокортикоидов приводит к гипонатриемии, гиперкалиемии, дегидратации и артериальной гипотензии. Эта форма проявляется в первые месяцы жизни, чаще у мальчиков. Без лечения, как правило, заканчивается летально. Сольтеряющая форма может развиваться в результате недостаточности и других ферментов.
• Недостаточность 18-оксидазы (*124080, кортикостерон метил оксидаза I, r ) проявляется изолированным нарушением синтеза альдостерона с развитием сольтеряющей формы заболевания. Больные умирают в раннем детстве.
• Недостаточность 20,22-десмолазы (*201710, r ) проявляется нарушением синтеза кортизола, альдостерона и андрогенов. Это приводит к потере солей, глюкокортикоидной недостаточности и задержке полового маскулинизирующего развития у плодов мужского пола. Развивается так называемая врождённая липоидная гиперплазия коры надпочечников. Больные погибают в раннем детстве.
Клиническая картина
• Вирильная форма проявляется главным образом избытком андрогенов •• У девочек часто наблюдают врождённые изменения наружных половых органов (пенисообразный клитор, урогенитальный синус, мошонкообразные большие половые губы). В постнатальном периоде вирилизация продолжается (рост мышечной массы по мужскому типу, грубый голос, гирсутизм, аменорея, атрофия грудных желёз) •• У младенцев мужского пола следствие избытка андрогенов во время развития плода — макрогенитосомия. В постнатальном периоде наступает преждевременное половое созревание на фоне недоразвития яичек (сперматогенез отсутствует).
• Сольтеряющая форма наблюдается обычно у новорождённых и детей первого года жизни. Проявляется срыгиванием, рвотой, диареей, похуданием, артериальной гипотензией и судорогами. Дефицит кортизола обычно не имеет значительных клинических проявлений, т.к. несмотря на недостаточность конкретного фермента, стимуляция АКТГ и гиперплазия надпочечников поддерживают уровень кортизола на нижней границе нормы.
• Гипертензивная форма. Наряду с вирилизацией у девочек и макрогенитосомией у мальчиков отмечается стойкая артериальная гипертензия.
Дифференциальная диагностика. Её проводят с надпочечниковой недостаточностью, гермафродитизмом другого генеза, различными вариантами преждевременного полового созревания, андроген-продуцирующей опухолью надпочечников. Сольтеряющую форму следует также дифференцировать с пилоростенозом, кишечными инфекциями и интоксикациями.
ЛЕЧЕНИЕ
Лекарственная терапия. ГК пожизненно (подавляют гиперпродукцию АКТГ, а также надпочечниковых андрогенов). При натрий-дефицитной форме может оказаться необходимой заместительная терапия минералокортикоидами (флудрокортизон). Симптоматическое лечение (восстановление концентрации хлорида натрия).
Хирургическое лечение. В первые несколько лет жизни проводят реконструктивную операцию на наружных половых органах девочек.
Синонимы • Врождённая дисфункция коры надпочечников • Синдром Апера–Галле • Синдром Крука–Апера–Галле.
Сокращение. 17-ОКС — 17-оксикортикостероиды.
Код вставки на сайт
Синдром адреногенитальный
Адреногенитальный синдром — врождённое патологическое состояние, обусловленное дисфункцией коры надпочечников с чрезмерной секрецией андрогенов и проявляющееся признаками вирилизации.
Этиология и патогенез. Синдром обусловлен недостаточностью одного из ферментов, необходимых для синтеза кортизола. Дефицит кортизола стимулирует выработку АКТГ, что приводит к гиперплазии коры надпочечников и избыточной продукции АКТГ-зависимых стероидов, синтез которых при данной недостаточности фермента не нарушен (в основном, надпочечниковых андрогенов — дегидроэпиандростерона, андростендиона и тестостерона).
Генетические аспекты
• Недостаточность 21-гидроксилазы (*201910, КФ 1.14.99.10, 6p21.3, мутации генов CYP21, CA2, CYP21P, r ) наблюдают в 95% случаев адреногенитального синдрома •• При умеренном дефиците фермента развивается вирильная (неосложнённая) форма синдрома, при которой придают значение исключительно симптомам избытка андрогенов (глюкокортикоидная и минералокортикоидная недостаточность не проявляется) •• При полном блоке фермента развивается сольтеряющая (тяжёлая) форма (синдром Дебре–Фибигера). Наряду с уменьшением синтеза кортизола снижена продукция альдостерона; дефицит минералокортикоидов приводит к гипонатриемии, гиперкалиемии, дегидратации и артериальной гипотензии. Эта форма проявляется в первые месяцы жизни, чаще у мальчиков. Без лечения, как правило, заканчивается летально. Сольтеряющая форма может развиваться в результате недостаточности и других ферментов.
• Недостаточность 18-оксидазы (*124080, кортикостерон метил оксидаза I, r ) проявляется изолированным нарушением синтеза альдостерона с развитием сольтеряющей формы заболевания. Больные умирают в раннем детстве.
• Недостаточность 20,22-десмолазы (*201710, r ) проявляется нарушением синтеза кортизола, альдостерона и андрогенов. Это приводит к потере солей, глюкокортикоидной недостаточности и задержке полового маскулинизирующего развития у плодов мужского пола. Развивается так называемая врождённая липоидная гиперплазия коры надпочечников. Больные погибают в раннем детстве.
Клиническая картина
• Вирильная форма проявляется главным образом избытком андрогенов •• У девочек часто наблюдают врождённые изменения наружных половых органов (пенисообразный клитор, урогенитальный синус, мошонкообразные большие половые губы). В постнатальном периоде вирилизация продолжается (рост мышечной массы по мужскому типу, грубый голос, гирсутизм, аменорея, атрофия грудных желёз) •• У младенцев мужского пола следствие избытка андрогенов во время развития плода — макрогенитосомия. В постнатальном периоде наступает преждевременное половое созревание на фоне недоразвития яичек (сперматогенез отсутствует).
• Сольтеряющая форма наблюдается обычно у новорождённых и детей первого года жизни. Проявляется срыгиванием, рвотой, диареей, похуданием, артериальной гипотензией и судорогами. Дефицит кортизола обычно не имеет значительных клинических проявлений, т.к. несмотря на недостаточность конкретного фермента, стимуляция АКТГ и гиперплазия надпочечников поддерживают уровень кортизола на нижней границе нормы.
• Гипертензивная форма. Наряду с вирилизацией у девочек и макрогенитосомией у мальчиков отмечается стойкая артериальная гипертензия.
Дифференциальная диагностика. Её проводят с надпочечниковой недостаточностью, гермафродитизмом другого генеза, различными вариантами преждевременного полового созревания, андроген-продуцирующей опухолью надпочечников. Сольтеряющую форму следует также дифференцировать с пилоростенозом, кишечными инфекциями и интоксикациями.
ЛЕЧЕНИЕ
Лекарственная терапия. ГК пожизненно (подавляют гиперпродукцию АКТГ, а также надпочечниковых андрогенов). При натрий-дефицитной форме может оказаться необходимой заместительная терапия минералокортикоидами (флудрокортизон). Симптоматическое лечение (восстановление концентрации хлорида натрия).
Хирургическое лечение. В первые несколько лет жизни проводят реконструктивную операцию на наружных половых органах девочек.
Синонимы • Врождённая дисфункция коры надпочечников • Синдром Апера–Галле • Синдром Крука–Апера–Галле.
Сокращение. 17-ОКС — 17-оксикортикостероиды.