АНОМАЛИЯ
Полезное
Смотреть что такое «АНОМАЛИЯ» в других словарях:
АНОМАЛИЯ — (греч. anomalia, от a отриц. част., n плавная буква, и homalos ровный). 1) неправильность; все, отступающее от обыкновенного порядка. 2) угол, на котором планета находится в точке дальнейшего расстояния от Солнца. Словарь иностранных слов,… … Словарь иностранных слов русского языка
АНОМАЛИЯ — (аномалия устар.), аномалии, жен. (греч. anomalia неравенство) (книжн.). Уклонение от закономерности явлений, отступление от существующего положения или порядка, неправильность. Аномалия в физическом развитии. Курская магнитная аномалия. (см.… … Толковый словарь Ушакова
АНОМАЛИЯ — (греческое anomalia), отклонение от нормы, от общей закономерности, неправильность … Современная энциклопедия
АНОМАЛИЯ — (греч. anomalia) отклонение от нормы, от общей закономерности, неправильность … Большой Энциклопедический словарь
АНОМАЛИЯ — АНОМАЛИЯ, и, жен. (книжн.). Отклонение от нормы, общей закономерности; неправильность. Магнитная а. | прил. аномальный, ая, ое. Толковый словарь Ожегова. С.И. Ожегов, Н.Ю. Шведова. 1949 1992 … Толковый словарь Ожегова
АНОМАЛИЯ — жен., греч. уклонение от обычного, несходство с обыкновенным, отступление в каком либо явлении природы; изъятие, исключение, уклонение, причуда, необычайность, странность. | астрах. угол расстояния планеты от афелия или перигелия. Аномальный,… … Толковый словарь Даля
АНОМАЛИЯ — (Anomaly) буквально: отступление от нормы. А. магнитная нарушение нормального распределения элементов земного магнетизма по земной поверхности. А. гравитационная нарушение нормального распределения сил тяжести по земной поверхности. А.… … Морской словарь
Аномалия — (греч.) так называется отступление или уклонение от правила,поэтому аномальным называют все, отступающее или уклоняющееся отправильного или нормального. А. в области природы считаются такиеявления, которые, вопреки законам природы, представляются … Энциклопедия Брокгауза и Ефрона
Научная электронная библиотека

Юров И. Ю., Воинова В. Ю., Ворсанова С. Г., Юров Ю. Б.,
5.1. Классификация врожденных аномалий
Различают изолированные аномалии (примеры, расщелина твердого нёба, врожденный дефект межжелудочковой перегородки сердца), системные (в пределах одной системы органов, например, скелетные дисплазии) и множественные (затрагивают две и более системы органов).
Изолированные врожденные аномалии. Среди изолированных врожденных аномалий различают мальформации, дизрупции, деформации и дисплазии (рис. 16). Последние правильнее отнести к системным аномалиям. Схема нарушений морфогенеза представлена на рис. 16.
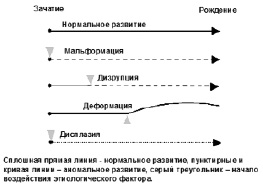
Рис. 16. Схема нарушений морфогенеза
Мальформация – это первичный структурный дефект органа или его части, который является результатом генетических нарушений. Мальформация подразумевает то, что раннее внутриутробное развитие определенной ткани или органа было нарушено. Частые примеры мальформаций – это врожденные пороки сердца, расщелина губы и неба, дефекты нервной трубки. Большинство мальформаций, затрагивающих только один орган, является мультифакторным признаком. Множественные мальформации наиболее часто возникают вследствие хромосомных аномалий.
Дизрупция – возникновение аномальной структуры органа или ткани в результате воздействия внешних факторов, нарушающих процесс нормального развития. Её вызывают такие внешние факторы, как ишемия, инфекция, травма и др. Примером дизрупции является нарушение развития конечностей плода вследствие приема препарата талидомида во время беременности. По определению дизрупция – негенетическое событие.
Деформация – результат механических воздействий, которые искажают морфологию плода. Например, дислокация бедра может быть вызвана недостатком амниотической жидкости (олигогидрамнион) или многоплодной беременностью, либо структурной аномалией матки. Деформации обычно возникают во второй половине беременности и имеют хороший прогноз при адекватном лечении.
Дисплазия – аномальная организация клеток в ткани. Например, при эктодермальной дисплазии вовлечены все ткани эктодермального происхождения – поражаются кожа, ногти, волосы и др. Большинство дисплазий вызываются моногенными дефектами и связаны с высоким риском рождения детей с данной аномалией в семье.
Множественные врожденные аномалии: секвенции, синдромы и ассоциации.
Секвенция – комплекс признаков, являющихся следствием каскада событий, инициированных одним фактором. Например, при секвенции Поттера, хроническое подтекание амниотической жидкости либо нарушение образования мочи ведет к олигогидрамниону, что, в свою очередь, ведет к компрессии плода, формированию «помятого» лица, дислокации бедер, пяток и гипоплазии легких, что может вызвать неонатальную гибель ребенка от дыхательной недостаточности (рис. 17).
Синдром – распознаваемый комплекс аномалий, этиология которых известна. Примерами являются хромосомные и моногенные синдромы. Диагностика синдромов значительно облегчается существованием электронных баз данных, таких как POSSUM (Pictures of Standard Syndromes and Undiagnosed Malformations), Лондонская база данных и др.
Ассоциация – понятие, которое возникло в связи с тем, что некоторые аномалии имеют тенденцию встречаться в сочетании друг с другом, а не случайно, и это необъяснимо с позиций секвенции или синдрома. Основным отличием ассоциации от синдрома является непостоянство набора аномалий у разных больных. Примером является VATER-ассоциация – комплекс из вертебрального, анального, трахео-эзофагального и ренального пороков. Ассоциации имеют низкий рекуррентный риск, и причина их не ясна.
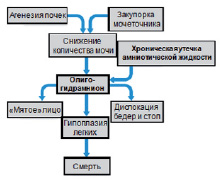
Рис. 17. Секвенция: каскад событий,
ведущий к уменьшению количества амниотической жидкости (олигогидрамниону)
Что касается структуры причин врожденных аномалий развития, то генетически обусловленные формы составляют 20–30 %, мультифакторные – 30–40 %, экзогенные – 2–5 %, неизвестной этиологии – 25–50 %.
АНОМАЛИЯ
Полезное
Смотреть что такое «АНОМАЛИЯ» в других словарях:
АНОМАЛИЯ — (греч. anomalia, от a отриц. част., n плавная буква, и homalos ровный). 1) неправильность; все, отступающее от обыкновенного порядка. 2) угол, на котором планета находится в точке дальнейшего расстояния от Солнца. Словарь иностранных слов,… … Словарь иностранных слов русского языка
АНОМАЛИЯ — (от греч. а – отриц. частица и nomos – закон) отклонение от нормы, от общей закономерности. Равнозначно: анормальность. Философский энциклопедический словарь. 2010. АНОМА … Философская энциклопедия
АНОМАЛИЯ — (аномалия устар.), аномалии, жен. (греч. anomalia неравенство) (книжн.). Уклонение от закономерности явлений, отступление от существующего положения или порядка, неправильность. Аномалия в физическом развитии. Курская магнитная аномалия. (см.… … Толковый словарь Ушакова
АНОМАЛИЯ — (греческое anomalia), отклонение от нормы, от общей закономерности, неправильность … Современная энциклопедия
АНОМАЛИЯ — (греч. anomalia) отклонение от нормы, от общей закономерности, неправильность … Большой Энциклопедический словарь
АНОМАЛИЯ — АНОМАЛИЯ, и, жен. (книжн.). Отклонение от нормы, общей закономерности; неправильность. Магнитная а. | прил. аномальный, ая, ое. Толковый словарь Ожегова. С.И. Ожегов, Н.Ю. Шведова. 1949 1992 … Толковый словарь Ожегова
АНОМАЛИЯ — жен., греч. уклонение от обычного, несходство с обыкновенным, отступление в каком либо явлении природы; изъятие, исключение, уклонение, причуда, необычайность, странность. | астрах. угол расстояния планеты от афелия или перигелия. Аномальный,… … Толковый словарь Даля
АНОМАЛИЯ — (Anomaly) буквально: отступление от нормы. А. магнитная нарушение нормального распределения элементов земного магнетизма по земной поверхности. А. гравитационная нарушение нормального распределения сил тяжести по земной поверхности. А.… … Морской словарь
Аномалия — (греч.) так называется отступление или уклонение от правила,поэтому аномальным называют все, отступающее или уклоняющееся отправильного или нормального. А. в области природы считаются такиеявления, которые, вопреки законам природы, представляются … Энциклопедия Брокгауза и Ефрона
Научная электронная библиотека

Юров И. Ю., Ворсанова С. Г., Воинова В. Ю., Чурносов М. И., Юров Ю. Б.,
4.2. Принципы клинической диагностики наследственных болезней
Диагностика наследственных заболеваний чрезвычайно сложна из-за их многочисленности (несколько тысяч нозологических форм) и низкой частоты (частота многих заболеваний составляет 1:200000 и более).
Термин синдром обычно применяют для обозначения совокупности воспроизводимых симптомов, имеющих общую этиологию. Однако в клинической генетике его используют для обозначения отдельных нозологических форм. Это связано с тем, что исторически многие из них были описаны как симптомокомплексы без понимания этиологии. Несмотря на то, что в дальнейшем стали известны генетические причины этих заболеваний, за ними традиционно сохранилось название «синдром». Таким образом, для наследственной патологии понятия «болезнь» и «синдром» равнозначны. Для обозначения некоторых нозологических форм употребляются оба термина (например, болезнь Дауна и синдром Дауна, болезнь Марфана и синдром Марфана).
Среди симптомов наследственных болезней редко встречаются патогномоничные. Чаще всего один и тот же признак встречается при различных заболеваниях. Так, катаракта и помутнение хрусталика наблюдаются при более чем 30 наследственных заболеваниях, а умственная отсталость при сотнях нозологических форм. Поэтому, в клинической генетике значим синдромальный подход к диагностике, согласно которому только сочетание симптомов является специфичным для конкретного заболевания.
На основе данных об особенностях клинических проявлений наследственной патологии строится схема обследования больного. Важным методом с точки зрения генетики является антропометрия. Необходимо оценивать следующие антропометрические показатели: длину и массу тела больного, тип телосложения, соотношение длины конечностей и туловища, а также отдельных частей конечностей (например, плеча и предплечья), окружности груди и головы, соотношение продольного и поперечного размера черепа, длину стопы и кисти. Эти данные оцениваются по специально разработанным центильным таблицам, основные из которых представлены в Приложении. При многих наследственных заболеваниях антропометрические показатели выходят за пределы допустимых вариаций вследствие нарушения роста скелета, диспропорциональности развития его отдельных частей. Например, высокий рост, длинные верхние и нижние конечности, пальцы могут быть признаками синдрома Марфана. Уменьшение окружности головы и размеров стоп помогают в диагностике синдрома Ретта.
Очень важно выявить у больного врождённые аномалии развития, которые могут быть либо проявлением наследственной патологии, либо следствием тератогенного воздействия средовых факторов. Следует остановиться на различиях между следующими употребляемыми в литературе терминами: врождённые пороки развития, врождённые аномалии, микроаномалии развития.
Врождённый порок развития – это морфологический дефект органа или его части, или большой области тела, ведущий к нарушению функции органа (органов) либо социальной адаптации индивидуума. Врождённая аномалия (или дефект) – более общее понятие, это любая функциональная или структурная аномалия, вызванная либо генетическим заболеванием, либо средовым событием, предшествующим рождению. Врождённые пороки развития подробно рассмотрены ниже (см раздел «Врождённые аномалии и синдромы», глава 6).
Микроаномалии развития (МАР) – необычные морфологические черты, которые, в отличие от пороков развития, не сопровождаются нарушениями функции органа или системы.
Как было сказано выше, при обследовании больных необходимо обращать внимание на МАР. Последние встречаются также и у здоровых людей, но наличие нескольких МАР указывает на высокую вероятность наследственной патологии. В таблицах 3 и 4 представлен перечень наиболее значимых микроаномалий и способы определения некоторых из них.
МАР органов или части тела
I. Лицевая область и остальная часть черепа
Лицо: плоское, круглое, треугольное, вытянутое, грубые черты, плоский профиль.
Брови: сросшиеся (синофриз), кустистые.
Нос: короткий, клювовидный, загнутый, седловидная переносица, широкая плоская переносица, плоские крылья носа, открытые вперёд ноздри.
Фильтр: длинный, короткий, плоский, глубокий.
Волосы: торчащие, два завитка (две макушки).
Голова: брахицефалия, долихоцефалия, тригоноцефалия, акроцефалия, выступающий или скошенный лоб, выступающий затылок, затылочная шпора (выступающая затылочная кость), плоский затылок.
II. Область глазных щелей и вокруг них
Веки: эпикант, телекант, птоз (малый), колобома век.
Глазные щели: узкие, короткие.
Разрез глазных щелей: монголоидный, антимонголоидный.
Расстояние между глазами: гипотелоризм, гипертелоризм.
Склеры: голубые, телеангиэктазии на склерах.
Радужная оболочка глаза: колобома, гетерохромия.
Ресницы: неправильный рост (например, двойной ряд ресниц).
III. Область ротовой полости и вокруг нее
Челюсти: прогения (выступающая нижняя челюсть), ретрогения (смещенная назад нижняя челюсть), прогнатия (выступающая верхняя челюсть), макрогения (большая нижняя челюсть) и микрогения (маленькая нижняя челюсть), микрогнатия (маленькая верхняя челюсть) и макрогнатия (большая верхняя челюсть).
Подбородок: скошенный, выступающий.
Язык: исчерченность, макроглоссия и микроглоссия (увеличение и уменьшение языка), короткая уздечка.
Губы: тонкие, толстые, с бороздами, с множественными уздечками.
Рот: макростомия (большой рот) и микростомия (маленький рот).
Нёбо: плоское, высокое, арковидное, готическое, раздвоение язычка.
Зубы: неправильное расположение, неправильная форма, врождённый избыток или врождённое отсутствие одного или нескольких зубов, гипоплазия эмали, диастема верхняя и нижняя (увеличенние промежутка между первыми резцами), тремы (широкие промежутки между зубами).
Асимметрия размера, большие, маленькие оттопыренные, косо направленные, низко расположенные, приросшая мочка, маленькая или отсутствующая мочка, уплощенные ушные раковины, неполное развитие завитка со сглаженным упрощенным рисунком, отсутствие козелка, околоушные придатки, преаурикулярная фистула.
V. Верхние конечности
Длина пальцев: брахидактилия (короткие пальцы), укорочение отдельных пальцев, арахнодактилия (длинные «паучьи» пальцы).
Количество пальцев: полидактилия (дополнительные пальцы), олигодактилия (уменьшение количества пальцев).
Форма пальцев: утолщение ногтевых фаланг, конусовидная форма.
Подвижность пальцев: камптодактилия (тугоподвижность межфаланговых суставов), сверхгибкость (гиперэкстензия).
Мизинец: короткий, серповидный (клинодактилия), дополнительные складки, отсутствие сгибательной складки либо одна складка.
Большой палец: широкий, трёхфаланговый, его гипоплазия.
Ногти: удвоение ногтя большого пальца, гипоплазия ногтей.
Синдактилия (сросшиеся пальцы). Поперечная ладонная борозда (четырехпальцевая борозда).
VI. Нижние конечности
Форма и длина: укороченные, удлинённые, вальгусная деформация (Х-образные) или варусная деформация (О-образные).
Форма стопы: полая, стопа-качалка.
Пальцы стоп: синдактилия, асимметрия длины пальцев, 3-й палец длиннее 2-го, глубокая борозда между 1-м и 2-м пальцами, сандалевидная щель, широкий большой палец, полидактилия, брахидактилия.
Глубокая складка на стопе.
VII. Область шеи и туловища
Шея: короткая, длинная, кривошея, крыловидные складки, низкая линия роста волос на шее.
Грудная клетка: воронкообразная, килевидная, щитовидная.
Соски: гипертелоризм, резкая гипоплазия, расположение на разном уровне, полителия (добавочные соски).
Пилонидальная ямка (втяжение кожи в виде ямки, часто локализуется в области крестца).
VIII. Половые органы
Шалевидная мошонка, увеличенный клитор.
IX. Кожа, ее придатки и подкожная клетчатка
Пятна на коже: гемангиомы, телеангиэктазии, крупные невусы (пигментированные выступающие участки кожи диаметром более 1 см), волосатый невус, пигментация, депигментация.
Повышенная растяжимость кожи, «лишняя» кожа.
Волосы: сухие, редкие, шерстистые, седая прядь надо лбом, «мыс вдовы», низкий рост волос на лбу, гипертрихоз (избыточный рост волос), гирсутизм (повышенное оволосение у девочек по мужскому типу), алопеция (облысение тотальное, гнёздное).
Ногти: короткие, широкие, вогнутые, гипоплазия, дистрофия ногтей, ногти в виде «часовых стекол».
Необычные ямки на лице и туловище, липомы, фибромы.
МАР, часто исчезающие в течение первых 2 лет жизни следующие: плоская капиллярная гемангиома на лице и шее, неполное развитие завитка уха, гипоплазия ногтя большого пальца ноги, широкая переносица, эпикант.
Способы определения некоторых микроаномалий
Определяется у больного в положении лежа на спине – затылок, шея и спина располагаются на одном горизонтальном уровне.
Гипер- и гипотелоризм глазных щелей
Определяется с помощью орбитального индекса (Ио): отношение расстояния между внутренними углами глазных щелей к окружности головы на уровне глаз.
где а – расстояние между внутренними углами глазных щелей;
b – окружность головы на уровне глаз.
Ио ≥ 7,5 % – гипертелоризм, Ио ≤ 5,2 % – гипотелоризм.
Смещение внутренних углов глазных щелей латерально при нормально расположенных глазницах.
Косое расположение ушных раковин
Угол, образованный линиями, проходящими вдоль ушной раковины и вертикалью, проходящей через мочку, > 20°.
Асимметрия ушных раковин
Различия в размерах правого и левого уха ≥ 15 %.
Низкое расположение ушных раковин
Определяется при сопоставлении верхней точки прикрепления ушной раковины с уровнем латерального угла глаза.
Верхний его край совпадает или ниже уровня середины 2-й фаланги 4-го пальца.
Расстояние между 1-м и 2-м пальцами нижних конечностей ≥ ширины 2-го пальца.
Сращение пальцев > 1/3 длины одного пальца.
Вычисляется на основании соскового индекса (Си) – отношения расстояния между центрами сосков и окружностью груди на уровне сосков (в %).
Врожденные заболевания детей (наследственные болезни)
Врожденные заболевания являются одной из распространенных причин младенческой и детской смертности, развития хронических болезней у детей и инвалидизации.
Быстрый переход
Врожденные заболевания могут быть обусловлены генетическими мутациями (передающимися по наследству или спонтанными), инфекциями матери во время беременности (цитомегаловирус, ветряная оспа, краснуха), воздействием лекарств и химических веществ, загрязняющих воздух, воду или пищу. Причины множества врожденных дефектов до сих пор неизвестны.
Генетическими или наследственными факторами обусловлены около 20 % врожденных заболеваний. К ним относятся нарушения, при которых мутация затрагивает один ген (серповидно-клеточная анемия); хромосомные нарушения, при которых хромосомы (или их части) отсутствуют (синдром Тернера) или имеют структурные изменения (увеличение количества хромосом или трисомия при синдроме Дауна); многофакторные нарушения, вызванные мутациями двух и более генов. Врожденные дефекты и нарушения развития могут вызывать делеции или дупликации отдельных генов (изменения митохондриальной ДНК). Примером такого заболевания является муковисцидоз, характеризующееся поражением экзокринных желез и жизненно-важных органов (легких и желудочно-кишечного тракта). Врожденные заболевания также ассоциированы со случайными (новыми) повреждениями генов, спонтанными (не наследующимися от родителей) мутациями (большинство случаев ахондроплазии).
По статистике, врожденные заболевания имеют 2–3 % младенцев. К возрасту 1 года их число увеличивается до 5 %, поскольку не все эти патологии диагностируются сразу после рождения.
Врожденные (наследственные) заболевания также классифицируют по типу наследования. При аутосомно-доминантном наследовании заболевание может передаваться от родителя к ребенку в 50 % случаев (мышечная дистрофия Дюшенна, хорея Гентингтона). При аутосомно-рецессивном наследовании генетическая аномалия передается ребенку только в том случае, если оба родителя наделены одним и тем же дефектным геном (муковисцидоз, серповидно-клеточная анемия). Здесь частота наследования составит 25 %, то есть в среднем у 1 ребенка из 4 детей этих родителей будет аутосомно-рецессивное заболевание.
Патологические состояния могут передаваться и при наследовании, сцепленном с полом (наследование гена, находящегося в половых хромосомах). Х-сцепленные рецессивные заболевания почти всегда ограничены мужским полом (гемофилия, дальтонизм, мышечная дистрофия Дюшенна). Х-сцепленные доминантные заболевания встречаются как у мальчиков, так и у девочек, однако у детей мужского пола имеют более тяжелое течение (Х-сцепленный гипофосфатемический рахит). Y-сцепленные заболевания встречаются довольно редко, поскольку Y-хромосома содержит всего несколько генов (ихтиоз).
Некоторые врожденные заболевания формально не относятся к генетическим, но имеют ту или иную выраженность наследования: наследуются факторы риска либо сам ген, но с низкой пенетрантностью (частотой проявления гена в признаках).
Гемофилия
Гемофилия — группа редких наследственных нарушений свертываемости крови, вызванных дефицитом необходимого белка (фактора свертывания крови).
Гемофилия A (классическая) встречается чаще (>80 % случаев) и связана с дефицитом VIII фактора свертывания крови, гемофилия B (болезнь Кристмаса) — реже (>10 % случаев), она обусловлена недостаточностью IX фактора свертывания крови. Гемофилия C встречается очень редко, обусловлена дефицитом XI фактора свертывания крови, чаще всего в классификации группы ее не упоминают.
Заболевание относится к X-сцепленным с полом, наследуется по рецессивному признаку по женской линии. Классическую гемофилию вызывают мутации гена F8, расположенного на X-хромосоме. Примерно в 70 % случаев заболевание наследуется по Х-сцепленному образцу, в остальных случаях оно возникает спонтанно (новая мутация), в дальнейшем спонтанное заболевание становится наследственным. Гемофилией A болеют практически исключительно мужчины, редкие случаи заболевания у женщин, носительниц дефектного гена, почти всегда характеризуются легким течением. Гемофилию B вызывают мутации гена F9, так же расположенного на X-хромосоме, она характерна для мужчин, женщины болеют очень редко. В некоторых случаях заболевание возникает спонтанно (приобретенная гемофилия A или B) и тоже связано с недостаточностью VIII или IX факторов свертывания крови. Приобретенная гемофилия является аутоиммунным заболеванием, при котором организм вырабатывает антитела, атакующие факторы свертывания крови (чаще всего VIII фактор). Примерно в половине случаев приобретенной гемофилии у пациента имеется связанное с ней основное состояние или заболевание (беременность, аутоиммунные заболевания, миелопролиферативные заболевания, воспалительные заболевания кишечника и др.), в остальных эпизодах причина остается невыясненной.
Гемофилия A затрагивает примерно 1 из 5 000 новорожденных мальчиков, гемофилия B — примерно 1 из 25 000. Около 60 % пациентов имеют тяжелую форму гемофилии, обычно им ставят диагноз при рождении или в течение первых 2 лет жизни.
Возраст манифестации гемофилии и тяжесть течения заболевания зависят от уровня активности факторов свертывания крови. Легкая форма характеризуется уровнем фактора свертывания крови (VIII или IX), превышающим 5 % от нормы, средняя — 1–5 %, тяжелая — ниже 1 %. У большинства пациентов, независимо от тяжести течения заболевания, эпизоды кровотечения чаще встречаются в раннем детском, детском и подростковом возрасте, чем в дальнейшей взрослой жизни.
При легкой и умеренной формах гемофилии длительные кровотечения могут возникнуть только в результате травмы, хирургического вмешательства или стоматологической процедуры. Нередко диагноз гемофилии ребенку устанавливают к 5–6 годам, обратив внимание на длительное посттравматическое кровотечение, длительное кровотечение во время стоматологического лечения или операции. К другим заметным симптомам легкого и умеренного течения гемофилии относятся непроходящие гематомы (синяки), частые носовые кровотечения, кровоточивость десен.
Для тяжелой формы гемофилии характерны эпизоды спонтанного кровотечения, которые приводят к кровоизлияниям различной локализации — в мягкие ткани, мышцы, суставы. При гемартрозах (кровоизлияние в полость сустава) возникает ограничение подвижности суставов, сопровождающееся острой болью и воспалением. У пациентов с тяжелой формой гемофилии спонтанные кровотечения чаще всего происходят в мышцы и суставы, однако могут затрагивать любой внутренний орган, включая почки, органы ЖКТ, головной мозг (гематурия, мелена, гематохезия, желудочно-кишечные кровотечения, внутричерепные кровотечения). Тяжелая форма гемофилии A обычно проявляется в раннем детском возрасте, диагноз чаще всего устанавливается к 2 годам ребенка. Обычными симптомами (при отсутствии лечения заболевания) здесь являются кровотечения из-за незначительных травм ротовой полости (прикусывание губ, языка, щек), подкожные гематомы, большие шишки после удара головой, спонтанные кровотечения (2–5 эпизодов в месяц).
Диагноз гемофилии A или B устанавливается на основании симптомов, истории личного и семейного анамнеза пациента, лабораторных исследований (общего анализа крови, коагулограммы, оценивающей состояние системы свертывания крови и активности ее белков, молекулярно-генетического исследования). Парам из группы риска по рождению ребенка с гемофилией необходимо получить генетическую консультацию на этапе планирования беременности. Определение конкретной мутации гена F8 (или F9) помогает не только выявить женщин-носительниц дефектного гена в конкретной семье, но и полезно для пренатальной диагностики гемофилии (амниоцентез, биопсия хориона).
Лекарств, воздействующих на причину гемофилии, не существует. Пациентам назначается заместительная терапия, направленная на восполнение дефицита белка (VIII или IX факторов свертывания крови). В идеале лечение проводится профилактически, с периодическим введением концентратов фактора свертывания крови через определенные промежутки времени (для предотвращения эпизодов кровотечения и связанных с ним осложнений, например, повреждения суставов). Возможно лечение «по требованию» (непосредственно при эпизодах кровотечения). В настоящее время авторитетными медицинскими организациями рекомендуется введение рекомбинантных факторов (они производятся в лаборатории и не содержат человеческих и животных белков) для снижения риска передачи различных вирусов при переливании компонентов крови.
Некоторым пациентам с легкой формой гемофилии назначается синтетический аналог вазопрессина — десмопрессин, повышающий VIII фактор свертывания крови (вводится внутривенно или интраназально), а также антифибринолитические средства, замедляющие распад факторов свертывания крови.
Примерно в 30 % случаев лечения тяжелых случаев гемофилии длительная заместитетельная терапия может приводить к изоиммунизации — образованию антител к вводимым факторам свертывания крови (иммунная система распознает вводимый фактор VIII как чужеродный). Этот процесс может сопровождаться аллергическими реакциями (разной степени тяжести), возрастает риск жизнеугрожающих кровотечений. В этом случае пациенту назначается альтернативное лечение — плазмаферез, иммунодепрессанты.
Гемохроматозы
Гемохроматоз — наследственное заболевание, характеризующееся нарушением обмена железа и его накоплением в тканях и органах.
Заболевание сопровождается повышенным всасыванием железа в желудочно-кишечном тракте и накоплением в печени, сердце, поджелудочной железе, суставах, гипофизе, что приводит к полиорганной недостаточности и развитию таких болезней, как цирроз печени, рак печени, диабет, болезни сердца и суставов.
Наследственный гемохроматоз вызывают мутации генов HFE, HFE2, HAMP, SLC40A1 и TfR2, он классифицируется в зависимости от возраста начала, генетической причины и способа наследования: гемохроматоз 1 типа, гемохроматоз 2 типа, гемохроматоз 3 типа, гемохроматоз 4 типа. Вторичный гемохроматоз ассоциирован с другими заболеваниями (не является наследственным) — анемией, хронической болезнью печени, инфекциями.
Гемохроматоз, редко возникающий в младенческом возрасте и не имеющий явной причины, называют неонатальным гемохроматозом. При этой форме заболевания избыточное накопление железа в тканях и органах начинается еще до рождения ребенка. Неонатальный гемохроматоз быстро прогрессирует и характеризуется поражениями печени, которые выявляют при рождении или в первые дни жизни. Дети с этим заболеванием часто рождаются недоношенными или имеют нарушения внутриутробного развития. Точная причина неонатального гемохроматоза неизвестна, есть версия, что он развивается в том случае, если иммунная система матери распознает клетки печени ребенка как чужеродные. Симптомы обычно включают гипогликемию, нарушения свертываемости крови, пожелтение кожных покровов и склер глаз, отеки. Диагноз устанавливается на основании признаков и симптомов, лабораторных и инструментальных исследований, биопсии печени. Лечение включает переливание крови, внутривенное введение иммуноглобулинов, трансплантацию печени.
Гемохроматоз 1 типа ассоциирован с мутациями в гене HFE, расположенном на коротком плече 6–й хромосомы, наследуется по аутосомно-рецессивному типу, является наиболее распространенным типом наследственного гемохроматоза и поражает в основном мужчин. Вероятность того, что ребенок унаследует мутацию в гене HFE от родителя составляет 25 %, вероятность того, что он станет носителем дефектного гена — 50 %.
Начальные симптомы заболевания обычно отмечаются в возрасте 40–60 лет и включают боль в животе, снижение полового влечения, усталость, слабость, боли в суставах, сухость кожи. В дальнейшем проявляются такие симптомы и осложнения, как изменение пигментации кожи (бронзовая кожа), выпадение волос на голове и туловище, аритмия, кардиомиопатия, хроническая сердечная недостаточность, сахарный диабет, гепатомегалия, цирроз печени, спленомегалия, атрофия яичек, аменорея (у женщин) и другие.
Диагноз устанавливается на основании симптомов заболевания, лабораторных исследований (уровень железа, ферритина, трансферрина, железосвязывающей способности сыворотки крови, маркеры функции печени, уровень глюкозы крови и др.), инструментальных исследований (рентгенография суставов, электрокардиография и эхокардиография сердца, УЗИ органов брюшной полости, МРТ печени и др.), биопсии печени, молекулярно-генетического исследования (в том числе для выявления родственников — имеющих гемохроматоз или носителей).
Лечение гемохроматоза направлено на удаление избытка железа из организма (флеботомия, хелатирование) с помощью забора крови (как при взятии анализов или донорстве крови, только в большем объеме) и специальных препаратов, образующих комплексное соединение с железом и способствующих его удалению из железосодержащих белков (дефероксамин).
Терапевтическая флеботомия сначала проводится 1–2 раза в неделю, поддерживающая — каждые 2–4 месяца. Вместе с этим проводится симптоматическое лечение сахарного диабета, патологий сердца, цирроза печени и других заболеваний, вызванных гемохроматозом. Пациентам запрещен прием препаратов железа и любых медикаментозных средств или комплексов, которые могут содержать железо и витамин C, запрещен алкоголь во избежание дальнейшего повреждения печени (если оно имеется), рекомендуется придерживаться диеты, исключающей продукты с высоким содержанием железа (красное мясо, яблоки, печень, шпинат).
Гемохроматоз 2 типа вызывают мутации в генах HFE2 или HAMP, он наследуется по аутосомно-рецессивному типу и проявляется чаще всего в детском возрасте. Для этого типа заболевания характерны следующие симптомы: высокие уровни ферритина и трансферрина, врожденный фиброз печени, артропатия, кардиомиопатия, генерализованная гиперпигментация кожи, мышечная слабость, гипогонадизм, задержка полового созревания, сахарный диабет, цирроз печени, остеопороз, спленомегалия, гепатомегалия и другие. Диагностика и лечение гемохроматоза 2 типа проводятся аналогично диагностике и лечению гемохроматоза 1 типа.
Гемохроматоз 3 типа вызывают мутации в гене TFR2, он наследуется по аутосомно-рецессивному типу. Симптомы обычно проявляются до 30 лет и, помимо вышеперечисленных, могут включать снижение уровня лимфоцитов и нейтрофилов в крови, красные или пурпурные пятна на коже. Диагностика и лечение гемохроматоза 3 типа проводятся аналогично диагностике и лечению гемохроматоза 1 и 2 типов.
Симптомы гемохроматоза 4 типа могут проявиться как в детском, так и во взрослом возрасте. Этот тип заболевания вызывают мутации гена SLC40A1, наследуется оно по аутосомно-доминатному типу. Вероятность того, что ребенок унаследует мутацию в гене SLC40A1 от родителя составляет 50 %, соответственно, вероятность того, что он станет носителем дефектного гена — тоже 50 %. Симптомы гемохроматоза 4 типа могут проявляться в виде повышенной утомляемости, слабости, болей в суставах, изменения пигментации кожи, затруднения дыхания, сердечной недостаточности, сахарного диабета, анемии, врожденного фиброза печени, цирроза печени, остеоартроза, катаракты. Диагностика и лечение гемохроматоза 4 типа проводятся аналогично диагностике и лечению гемохроматоза 1, 2 и 3 типов.
Наследственный гемохроматоз — заболевание с пониженной пенетрантностью (вероятностью того, что ген будет иметь любые фенотипические проявления) дефектного гена, т. е. у некоторых людей с мутациями гена HFE никогда не проявятся симптомы, при этом у их детей или других членов семьи с мутацией гена произойдет манифестация заболевания.
Мышечная дистрофия Дюшенна
Мышечная дистрофия Дюшенна — редкое наследственное заболевание, характеризующееся нарастающей мышечной слабостью с последующей атрофией мышц. Ассоциировано с мутацией гена DMD, расположенного на половой X-хромосоме (X-сцепленный рецессивный тип наследования).