Что такое auc в фармакологии
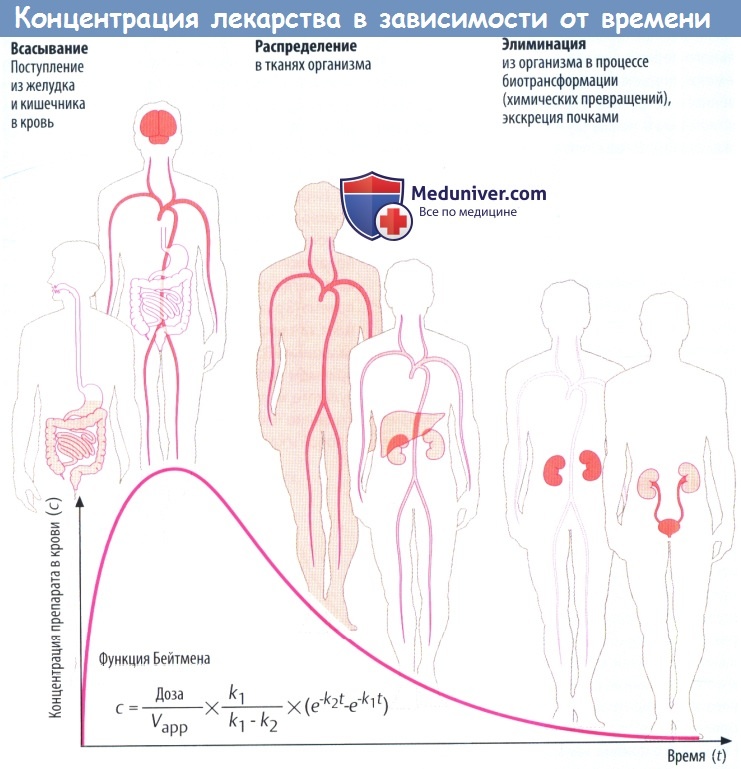
(А) Лекарственные средства попадают в организм и выводятся из него разными путями. Таким образом, организм представляет собой открытую систему, в которой фактическая концентрация препарата отражает взаимодействие между его поступлением (приемом) и эвакуацией (элиминацией).
Скорость всасывания препарата в желудке и кишечнике зависит от множества факторов: скорости растворения вещества (в случае приема твердой лекарственной формы) и транзита по ЖКТ, проницаемости слизистой для препарата, его градиента концентрации на границе слизистой и крови,кровоснабжения слизистой оболочки.
Всасывание из кишечника приводит к повышению концентрации лекарственного вещества в крови. Препарат разносится с кровью к различным органам (распределение), которые поглощают его в количестве, соответствующем его химическим свойствам и скорости кровотока через орган.
Например, органы с хорошим кровоснабжением, такие как головной мозг, получают большее количество препарата, чем органы с низким кровоснабжением. В результате поглощения тканями происходит снижение концентрации лекарственного вещества в крови. По мере снижения градиента на границе слизистой оболочки и крови всасывание в кишечнике замедляется. Пик концентрации в крови достигается тогда, когда количество вещества, покидающего кровь за единицу времени, равно количеству всосавшегося.
Поступление вещества в ткани печени и почек представляет собой перемещение в органы выведения. Концентрация препарата в крови в различные периоды времени представляет собой совокупность процессов абсорбции, распределения и элиминации, которые пересекаются во времени.
Если распределение происходит значительно быстрее, чем элиминация, снижение концентрации в крови вначале происходит быстро, а затем замедляется. Фаза быстрого снижения обозначается как α-фаза (фаза распределения), медленного — как β-фаза (фаза элиминации). Если препарат распределяется быстрее, чем абсорбируется, концентрацию препарата в крови можно описать математически упрощенной функцией Бейтмена (k1 и k2 — константы скорости для абсорбции и элиминации соответственно).
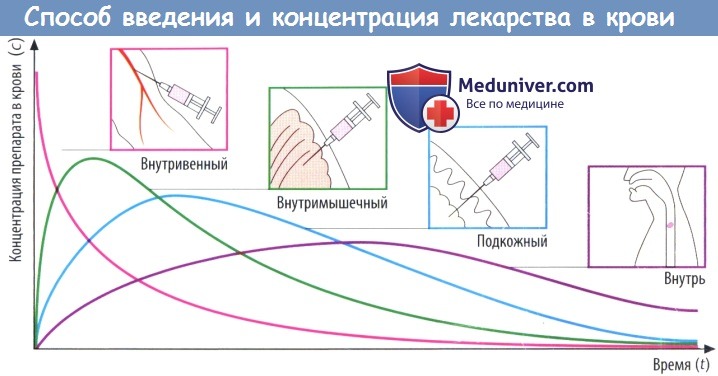
(В) Скорость абсорбции зависит от способа введения препарата. Чем выше скорость абсорбции, тем короче будет время (tmax), которое требуется для достижения пика концентрации в плазме (cmax), тем выше будет cmax и тем раньше уровень препарата в крови снова начнет снижаться.
Площадь под кривой, описывающей зависимость концентрации препарата в крови от времени (AUC), не зависит от пути введения препарата при условии, что доза и биодоступность остаются теми же (закон соответственных состояний). Таким образом, AUC можно использовать для вычисления биодоступности (F) препарата.
Значение AUC, измеренное после приема внутрь и в/в введения определенной дозы конкретного лекарственного вещества, соответствует проценту вещества, попавшего в системный кровоток после приема внутрь: F = AUCприем внутрь/AUCв/в введение.
Определение концентрации препарата в крови позволяет сравнить различные патентованные лекарственные средства, содержащие одно и то же действующее вещество в одинаковой дозе. Идентичные кривые зависимости концентрации в крови от времени для препаратов различных производителей (при условии стандартных лекарственных форм) означают биоэквивалентность стандартного вещества и нового исследуемого препарата.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Что такое auc в фармакологии
AUC — аббревиатура от англоязычного Area Under the Curve (площадь под кривой). В медицинской и фармацевтической обычно используется без перевода.
AUC в фармакокинетике
AUC — фармакокинетический параметр, характеризующий суммарную концентрацию лекарственного препарата в плазме крови в течение всего времени наблюдения. Математически определяется как интеграл от 0 до ∞ функции концентрации препарата (фармакокинетической кривой) в плазме крови от времени и равен площади фигуры, ограниченной фармакокинетической кривой и осями координат. Параметр AUC связан с другими фармакокинетическими параметрами — объемом распределения, общим клиренсом. При линейности кинетики препарата в организме величина AUC пропорциональна общему количеству (дозе) препарата, попавшего в системный кровоток.

AUC T — площадь под частью фармакокинетической кривой, от начала исследования (t = 0) до некоторого времени t = T (обычно заданное в часах). Например, AUC24 равен площади под фармакокинетической кривой в течение первых 24-х часов исследования.
AUC в исследовании кислотности органов желудочно-кишечного тракта
AUC [H + ] или просто AUC («интегральная кислотность») — широко используемое в зарубежных работах обозначение показателя, применяемого для оценки кислотоподавляющих свойств лекарственных препаратов и равного площади под заданным участком рН-метрической кривой. В отечественной литературе этот показатель называется «площадь ощелачивания» (Рапопорт С.И. и др.). AUC [H + ] равен S (iн, iк, 0) работы «Математический анализ компьютерных рН-грамм верхних отделов ЖКТ (п. 4 на стр. 2)».
Основные фармакокинетические параметры

Фармакокинетика — раздел клинической фармакологии, изучающий пути введения, биотрансформацию, связь с белками крови, распределение и выведение лекарственных средств (ЛС).
Один из основных показателей, определяющих фармакологический эффект, — концентрация ЛС в области рецептора, однако в условиях целостного организма установить её невозможно. Экспериментально доказано, что в большинстве случаев имеется корреляция между концентрацией препарата в крови и его содержанием в других биологических жидкостях и тканях.
Поэтому для определения фармакокинетических параметров ЛС изучают его содержание в крови. Чтобы получить соответствующие представления о поступлении препарата в кровь и выведении его из организма, определяют содержание ЛС в плазме крови в течение длительного времени, используя методы жидкостной или газожидкостной хроматографии, радиоиммунный и иммуноферментный анализы, спектрофотометрический метод. На основании полученных данных строят график (фармакокинетическую кривую), отмечая на оси абсцисс время исследования, а на оси ординат — концентрацию ЛС в плазме крови.
В связи со сложностью описания деталей процесса распределения ЛС во всех органах и тканях, организм условно представляют в виде одной или нескольких изолированных проницаемой мембраной частей (камер), в которых Л С распределяется. Этот вид моделирования называют камерным. За центральную камеру обычно принимают кровь и хорошо кровоснабжаемые органы (сердце, лёгкие, печень, почки, эндокринные железы), за периферическую — менее интенсивно кровоснабжаемые органы и ткани (мышцы, кожу, жировую ткань). В этих камерах ЛС распределяется с разной скоростью: быстрее — в центральной, медленнее — в периферической. К наиболее простым относят однокамерную модель, когда предполагают, что после введения препарата его концентрация убывает по моноэкспоненциальному закону. В соответствии с законами линейной кинетики скорость изменения количества препарата в камере пропорциональна его количеству в этой камере.
Кажущийся объём распределения (Vd) — гипотетический объём жидкости организма, необходимый для равномерного распределения всего количества ЛС (введённой дозы) в концентрации, аналогичной таковой в плазме крови. Этот показатель измеряют в л/кг. При внутривенном введении объём распределения равен отношению дозы ЛС к его начальной концентрации в крови.
• Высокие значения объёма распределения свидетельствуют о том, что ЛС активно проникает в биологические жидкости и ткани. При этом, если ЛС активно связывается, например, жировой тканью, его концентрация в крови может практически мгновенно стать очень низкой, а объём распределения достигнет нескольких сотен литров, превысив реальный объём жидкостей организма. Поэтому этот показатель и называют кажущимся объёмом распределения.
• Объём распределения зависит от различных факторов.
— Физико-химические свойства ЛС (молекулярная масса, степень ионизации и полярности, растворимость в воде и жирах) влияют на его прохождение через мембраны.
— Физиологические факторы (возраст, пол, общее количество жировой ткани в организме). Например, у пожилых людей и ново рождённых Vd снижен.
— Патологические состояния, особенно заболевания печени, почек, сердечно-сосудистой системы (ССС).
Максимальная концентрация (Сmax) и время наступления максимальной концентрации (Тmax). При поступлении ЛС в системный кровоток (в случае внесосудистого введения) его концентрация постепенно возрастает, достигая значения (Сmax) в момент Тmax, а затем начинает снижаться.
• Если процесс абсорбции имеет линейный характер (скорость процесса прямо пропорциональна количеству ЛС в системе), скорость этого процесса характеризуется константой абсорбции (kabs), измеряемой в часах и рассчитывается через период полувсасывания (Т1/2abs) — время, в течение которого всасывается 1/2 введённой дозы препарата.
Биодоступность (F) — часть дозы Л С (в %), достигшая системного кровотока после вне-сосудистого введения (в этом случае не всё количество препарата достигает системного кровотока).
• Абсолютную биодоступность определяют соотношением значений площади под кинетической кривой (area under curve, AUC) при вне-сосудистом и внутривенном введениях препарата.
— В рамках однокамерной модели при внутривенном введении площадь под кинетической кривой определяется отношением начальной концентрации в крови (Со) к константе элиминации (кеl)
— AUC прямо пропорциональна однократной дозе ЛС, введённой внутривенно (в/в), и обратно пропорциональна общему клиренсу препарата. Она связана с величиной объёма распределения:
где Vd — объём распределения, кеl — константа элиминации, D — доза, AUC — площадь под кинетической кривой.
• Биоэквивалентность (относительная биодоступность) — соотношение количества ЛС, поступившего в системное кровообращение при применении его в различных лекарственных формах или лекарственных препаратах, выпускаемых различными фирмами. Если сравниваемые ЛС аналогичны (действующее вещество, доза, лекарственная t форма), но изготовлены разными производителями, их называют дженериками, и в этом случае необходимо исследование их биоэкви— валентности. Два лекарственных препарата биоэквивалентны, если они обеспечивают одинаковую биодоступность ЛС.
Константа скорости элиминации (кеl) — процент снижения концентрации вещества в крови в единицу времени (отражает долю препарата, выводимую из организма за единицу времени). Элиминация складывается из процессов биотрансформации и экскреции. Константа скорости элиминации характеризует элиминацию в рамках однокамерной модели при линейном характере процесса выведения. Период полувыведения (Т1/2) — время, необходимое для снижения концентрации препарата в крови на 50% в результате элиминации. В рамках линейной модели Т1/2 рассчитывают по формуле:
• Практически за один Т 1 / 2 из организма выводится 50% ЛС, за два периода — 75%, за 3 периода — приблизительно 90% и т.д.
• Зависимость между Т1/2 и кеl важна для подбора режима дозирования и особенно для определения интервала между дозами.
Клиренс (CI) — объём плазмы или крови, полностью освобождающийся от ЛС в единицу времени. Этот показатель количественно характеризует выведение препарата и выражается в мл/мин или л/ч. В рамках линейной модели клиренс рассчитывают по формуле:
• Общий клиренс представляет собой сумму почечного и печёночного клиренсов (так как эти органы служат основными путями выведения ЛС). (Другие пути выведения или внепечёночный метаболизм при расчёте общего клиренса обычно не учитывают.)
— Печёночный клиренс характеризует биотрансформацию ЛС в печени (метаболический клиренс) и выведение с жёлчью (жёлчный клиренс).
— Почечный клиренс отражает выведение препарата с мочой. На пример, почечный клиренс циметидина приблизительно составляет 600 мл/мин, метаболический — 200 мл/мин, жёлчный — 10 мл/мин, поэтому общий клиренс равен 810 мл/мин.
• Основные физиологические факторы, определяющие клиренс, — функциональное состояние основных физиологических систем организма, объём притекающей крови и скорость кровотока в органе. Печёночный клиренс зависит от скорости печёночного кровотока или функциональной способности метаболизирующих ферментов. Например, клиренс лидокаина, интенсивно метаболизируемого печёночными ферментами, зависит прежде всего от скорости его доставки к печени (т.е. от объёма притекающей крови и скорости кровотока), поэтому, например, при застойной сердечной недостаточности он снижен. Клиренс же фенотиазинов зависит в основном от активности метаболизирующих ферментов, поэтому при поражении гепатоцитов клиренс препаратов этой группы резко снижается, вследствие чего концентрация их в крови значительно возрастает.
Равновесная (или стационарная) концентрация (Css) — концентрация, достигнутая при состоянии, когда в каждом интервале между приёмом очередных доз количество всасывающегося ЛС равно количеству элиминируемого [т.е. при стационарном (steady state), или равновесном, состоянии]. Т.е. если ЛС вводят в постоянной дозе через фиксированные интервалы времени, продолжительность которых меньше времени элиминации, его концентрация в крови возрастает, а затем колеблется в пределах средней величины между максимальными и минимальными значениями.
• При достижении С проявляется в полном объёме клинический эффект ЛС. Чем меньше Т1/2 ЛС, тем скорее достигается Си и тем выражение будут её колебания. Например, Т1/2 новокаинамида равен 2— 3 ч, и при назначении через каждые 6 ч его Css характеризуется большим разбросом значений. Поэтому для предупреждения и уменьшения колебаний Css в крови всё большее распространение получают лекарственные формы с замедленным высвобождением активного вещества.
В клинической практике фармакокинетические параметры используют, в частности, для расчёта назначаемых доз препаратов.
• Для расчёта нагрузочной дозы, требуемой для достижения необходимой эффективной концентрации ЛС в крови, используют объём распределения:
где Dнагр — нагрузочная доза, VD — объём распределения, С — концентрация ЛС в плазме крови.
• Для расчёта поддерживающей дозы, т.е. дозы, необходимой для поддержания нужной концентрации ЛС в крови, используют значение клиренса:
где Dnoд — поддерживающая доза, Сl — общий клиренс, Сss — равновесная концентрация.
К основным фармакокинетическим процессам относят всасывание, метаболизм (биотрансформацию), распределение и выведение ЛС.
Биодоступность пероральных препаратов
*Пятилетний импакт фактор РИНЦ за 2020 г.
Читайте в новом номере
В условиях интенсивного развития фармацевтической промышленности и огромного разнообразия препаратов для перорального приема у специалистов возникает необходимость обновлять свои знания о препаратах этой категории и процессах, происходящих в организме при их всасывании.
Современные фармацевтические технологии позволяют изменять в определенном диапазоне фармакокинетические параметры перорально принимаемого лекарственного средства. Как правило, эти технологии направлены на повышение биодоступности лекарственного средства и/или уменьшение риска возникновения нежелательных реакций. Объективной характеристикой количества всосавшегося вещества является площадь под кривой концентрация–время (AUC).
На основные фармакокинетические параметры перорально принятого препарата (максимальная концентрация, время ее достижения, период полувыведения, константа скорости элиминации и др.), кроме его физико–химических свойств, существенное влияние могут оказывать состояние желудочно–кишечного тракта пациента и физиологические процессы в системе пищеварения.
В связи с этим представляется важным рассмотреть факторы, влияющие в организме человека на биодоступность лекарственной формы при пероральном приеме.
Физиологические процессы в ЖКТ, влияющие на биодоступность
пероральных лекарственных форм
При пероральном приеме активное вещество таблетки (пока она не растворилась) проходит последовательно ротовую полость, пищевод, желудок, тонкий кишечник.
В ротовой полости таблетка обволакивается слюной. Многие лекарственные формы для перорального приема покрыты специальной оболочкой, препятствующей воздействию на них ферментов слюны, поэтому препараты, назначаемые перорально, не рекомендуется разжевывать.
Длина тонкой кишки – 5 м (двенадцатиперстной – 27–30 см). Пища находится в желудке от 30 мин. до полутора часов, в тонкой кишке – около 4 часов. Как правило, те же самые временные промежутки сохраняются и для лекарственных препаратов, принятых через рот.
Процесс усвоения некоторых лекарственных веществ начинается уже в желудке. Играет роль не только кислотность желудочного сока, но и время опорожнения желудка. У больных с высокой кислотностью желудочного сока вследствие спазма пилорического отдела замедляется опорожнение желудка, в результате чего всасывание лекарственных средств также замедляется. При анацидном состоянии опорожнение желудка наступает быстро, и это приводит к ускорению всасывания лекарственных средств и более быстрому наступлению фармакодинамического эффекта.
Из желудка лекарственное средство поступает в двенадцатиперстную кишку, куда открывается общий желчный проток и проток поджелудочной железы. Компоненты желчи способствуют растворению липофильных препаратов, оболочек, капсул, таблеток с кишечнорастворимым покрытием. В кишечнике активное вещество высвобождается из лекарственной формы и взаимодействует с кишечным соком. При этом соли желчных кислот могут образовывать с некоторыми лекарственными средствами нерастворимые комплексы, что приводит к снижению их биодоступности.
Большинство перорально принимаемых веществ всасывается в тонком кишечнике, имеющем чрезвычайно развитую поверхность (около 200 м2). Скорость поступления в системный кровоток при этом зависит от кровоснабжения кишечника в зоне всасывания.
На процесс всасывания лекарственных веществ существенное влияние оказывает пища. Для большинства лекарственных средств характерно замедление всасывания под влиянием пищи, связанное с замедлением опорожнения желудка. Особенно замедляет эвакуацию желудочного содержимого горячая, кислая, жирная, чрезмерно соленая или сладкая пища, а также пища густой консистенции. Но в некоторых случаях длительное пребывание лекарственных средств в желудке, способствует их более полному растворению и после перехода химуса в тонкую кишку биодоступность может повыситься (например, нитрофурантоин, гипотиазид). В связи с этим прием лекарственных препаратов связывают с режимом питания [1].
Во–первых, пища может выступать в качестве механического барьера, препятствующего контакту лекарственного средства с эпителием кишечника. Во–вторых, ряд продуктов могут оказывать влияние на рН содержимого желудка. В–третьих, пища может взаимодействовать с лекарственными средствами с образованием хелатных комплексов.
Препарат рекомендуется принимать до еды, если нужно быстро создать высокую концентрацию в крови. В остальных случаях считается целесообразным назначать лекарственные препараты после еды. Лекарственные средства, характеризующиеся значительной биотрансформацией при первом прохождении через печень, целесообразно принимать сразу после еды, при этом их биодоступность увеличивается за счет уменьшения пресистемной элиминации.
Следует отметить, что снижение биодоступности при приеме с пищей некоторых лекарственных препаратов не считают показанием к их назначению перед едой, так как при этом лекарственное средство может оказать раздражающее действие, вызвать обострение гастрита, язвенной болезни и способствовать развитию диспептических явлений.
Учитывая особенности фармакокинетики витаминов, их целесообразно принимать во время еды.
Энтеральный (пероральный) путь введения лекарственного средства является самым распространенным в практической медицине.
Он наиболее удобен и относительно безопасен для пациента. Однако для самого препарата это наиболее долгий и трудный путь, в результате которого происходят естественные потери самого активного вещества, достигающего рецепторного аппарата. В связи с этим некоторые вещества не имеют лекарственной формы для приема внутрь, так как они разрушаются ферментами желудочно–кишечного тракта (например, инсулин и другие белки), кислой средой желудка (например, бензилпенициллин).
Механизмы всасывания
Самый простой механизм транспорта лекарственных веществ – пассивная диффузия через мембраны клеток кишечной стенки (энтероцитов). Скорость всасывания в этом случае пропорциональна градиенту концентрации веществ и существенно зависит от их растворимости в мембране (наиболее легко таким путем всасываются липофильные неполярные вещества). Диффузии, как правило, подвергаются электролиты, находящиеся в недиссоциированном состоянии. Растворимость и степень ионизации лекарственного средства определяются рН содержимого желудка и кишечника. Необходимо подчеркнуть, что лекарственные средства путем пассивной диффузии хорошо всасываются не только в тонкой, но и толстой, и прямой кишках, что служит основой для разработки многих лекарственных средств с замедленным выделением действующего вещества, а также введения лекарственных средств ректальным путем.
Вода, электролиты и малые гидрофильные молекулы (например, мочевина) транспортируются в кровь другим механизмом – фильтрацией через поры в эпителии кишечника.
Активный транспорт, использующий специализированные механизмы клеточных мембран и молекулы–переносчики, обеспечивает всасывание гидрофильных полярных молекул, неорганических ионов, аминокислот, пиримидинов. Он требует для своего осуществления затрат энергии и характеризуется избирательностью, насыщаемостью, возможностью транспорта против градиента концентрации. При активном транспорте часто наблюдается конкуренция веществ за общий транспортный механизм (например, при усвоении некоторых витаминов и минеральных веществ). Степень всасывания зависит от дозы препарата, так как возможен феномен «насыщения белков–переносчиков».
Основной механизм всасывания ксенобиотков (синтезированных) лекарственных веществ – пассивная диффузия, активный транспорт используется редко. Для веществ природного происхождения, таких как аминокислоты, витамины, эссенциальные микроэлементы и др., в организме имеются специализированные активные транспортные механизмы. В этом случае основной путь усвоения – активный транспорт, а пассивная диффузия начинает играть роль только при очень высоких концентрациях.
Лекарственные вещества с большими молекулами или комплексы лекарственного вещества с крупной транспортной молекулой всасываются путем пиноцитоза. При этом происходит инвагинация мембраны клетки кишечного эпителия и образование пузырька (вакуоли), заполненного захваченной жидкостью вместе с лекарством. Вакуоль мигрирует по цитоплазме клетки к противоположной стороне и освобождает содержимое во внутреннюю среду организма. Однако пиноцитоз не имеет существенного значения для всасывания лекарственных средств и используется лишь в редких случаях (например, при усвоении комплекса цианокобаламина с белком – внутренним фактором Кастла) [1,2].
Фильтрация через поры имеет значение для всасывания лекарственных средств с молекулярной массой менее 100 Да.
Современные технологии
управляемого высвобождения
в производстве лекарственных средств
Современные аналитические методы позволяют определять в плазме крови сверхнизкие концентрации исследуемых лекарственных веществ, что дает возможность строить фармакокинетическую кривую с большой точностью и, соответственно, с большей определенностью судить о ее параметрах. Это в сочетании со знанием механизма усвоения конкретного вещества при пероральном приеме позволяет целенаправленно разрабатывать его лекарственную форму.
Для пероральных таблетированных препаратов применяются такие технологические приемы, как:
– использование вспомогательных веществ;
– гранулирование;
– микрокапсулирование;
– применение специального прессования;
– покрытие оболочками и т.д.
С их помощью можно изменять время распада таблетки, скорость растворения или выделения лекарственного вещества, место выделения и длительность нахождения в определенной зоне желудочно–кишечного тракта (над окном всасывания). А это, в свою очередь, определяет скорость и полноту всасывания, динамику концентрации лекарственного вещества в крови, то есть биодоступность препарата [3].
К сожалению, большинство применяемых в фармацевтике технологий производства таблетированных препаратов не позволяют независимо влиять на время и на место усвоения активного вещества, так как обычно препарат непрерывно продвигается по желудочно–кишечному тракту вместе с пищевым комком или химусом. То есть задержка времени высвобождения активного вещества неизбежно сдвигает место высвобождения ниже по пищеварительному тракту. Для некоторых конкретных препаратов предлагаются оригинальные методы решения этой проблемы: таблетки из микрочастиц с адгезивными свойствами, которые «приклеиваются» к слизистой оболочке, или таблетки, разбухающие в желудке настолько, что плавают на поверхности и/или не могут пройти через пилорический сфинктер в кишечник [4].
На скорость распада таблетки в желудке влияет способ их производства. Так, обычные (прессованные) таблетки прочнее тритурационных (формованных). Скорость распада зависит и от вспомогательных веществ, используемых для придания необходимых свойств таблетируемой смеси (сыпучесть, пластичность, прессуемость, содержание влаги и т.д.).
Кишечнорастворимые таблетки получают путем покрытия их желудочно–резистентной оболочкой или прессованием гранул или микрокапсул, предварительно покрытых такими оболочками. При необходимости оболочки могут обеспечивать и более длительную задержку растворения, чем на 1 час, который таблетка проводит в желудке.
Оболочка может быть достаточно толстой, например, сахарной, которая иногда имеет большую массу, чем ядро таблетки, содержащее лекарственное вещество. Тонкие пленочные оболочки (менее 10% от массы таблетки) могут выполняться из целлюлозы, полиэтиленгликолей, желатина, гуммиарабика и т.д.
Подбором оболочки и введением дополнительных веществ можно достичь замедления нарастания концентрации активного вещества в крови, что важно для снижения риска развития нежелательной реакции и/или сдвинуть время достижения максимума на несколько часов, если требуется продлить действие препарата и тем самым сократить кратность приема в целях повышения комплаентности.
Таблетки пролонгированного действия (ретард), например, обычно получают прессованием микрогранул лекарственного вещества в биополимерной оболочке или распределеннием в биополимерной матрице. При постепенном (послойном) растворении основы или оболочки высвобождаются очередные порции лекарственного вещества.
Современные высокотехнологичные способы доставки позволяют достичь постепенного равномерного высвобождения лекарственного вещества. Например, за счет создания осмотического давления внутри капсулы с действующим веществом. На этом принципе созданы новые лекарственные формы известных препаратов нифедипина (Procardia XL, Pfeizer), оксибутина хлорида (Ditrophan XL, Ortho–McNeil), метилфенидата (Concerta, ALZA) [5,6].
Управляемое (контролируемое) высвобождение может достигаться использованием в таблетках микрокапсул с лекарственным веществом, покрытых специальным полимером. После растворения внешнего слоя внутрь капсулы начинает поступать жидкость и по мере растворения ядра происходит постепенное высвобождение и диффузия лекарственного вещества через мембрану капсулы [7].
Основным фактором, ограничивающим производство и использование подобных лекарственных форм, остается условие необходимости высвобождения всего действующего начала за время прохождения таблеткой основных мест всасывания лекарственных средств в желудочно–кишечном тракте – 4–5 часов.
Проблемы использования технологий управляемого высвобождения для производства комбинированных
препаратов
Особые технологические проблемы ставят перед разработчиками комбинированные препараты, содержащие несколько активных веществ, требующих для оптимального всасывания различных условий.
Разумеется, если требования к месту и времени усвоения для компонентов одинаковы, можно просто таблетировать смесь или при необходимости (например, для ограничения контакта между компонентами при хранении) предварительно гранулировать и капсулировать компоненты.
Если компонентам требуются различные отделы ЖКТ для оптимального всасывания (желудок и тонкий кишечник или проксимальные и дистальный отделы тонкого кишечника), то таблетки прессуют из гранул с разными скоростями растворения. В этом случае возможно также использование технологий многослойного таблетирования или контролируемого высвобождения (с несколькими компартментами).
Если компоненты комплексного препарата должны усваиваться в разное время (но в одном месте желудочно–кишечного тракта), то альтернативы раздельному приему нет. Примером могут служить некоторые пероральные контрацептивы.
Обычно в состав комбинированного лекарственного средства не включают компоненты, отрицательно влияющие на сохранность, усвоение или фармакологическое действие друг друга. С витаминно–минеральными комплексами дело обстоит гораздо сложнее. Многие из них содержат в одной таблетке десятки компонентов, между которыми возможны описанные антагонистические взаимодействия. Закономерны следующие вопросы. Насколько целесообразно объединение в одной таблетке такого большого количества биологически активных веществ? Могут ли современные фармацевтические технологии создать такую лекарственную форму, которая обеспечила бы оптимальное всасывание всех компонентов при одновременном приеме?
Особенности всасывания витаминов
и микроэлементов
Рассмотрим особенности всасывания витаминов в желудочно–кишечном тракте (табл. 1).
Все витамины подразделяются на два класса в зависимости от их растворимости: жирорастворимые (липофильные) и водорастворимые (гидрофильные). К первым относятся витамины A, D, E и K, ко вторым – все витамины группы B, витамины С и H (биотин). Естественно, что растворимость существенно влияет на всасывание.
Жирорастворимые витамины могут перейти в водную среду лишь в составе мицелл, образующихся при эмульгировании желчью (солями желчных кислот) жиров в проксимальном отделе тонкого кишечника. Там же происходит всасывание этих витаминов, т.е. их освобождение из мицелл внутрь клеток кишечной стенки (энтероцитов), транспорт особыми гликопротеинами (хиломикронами) из цитоплазмы энтероцитов в лимфу и кровь. Всасывание жирорастворимых витаминов происходит в основном путем пассивной диффузии и зависит от наличия жиров в химусе.
При всасывании водорастворимых витаминов пассивная диффузия играет заметную роль только при приеме нагрузочных (высоких) доз. При приеме витаминных комплексов, содержащих компоненты в профилактических дозах, основное значение имеет активный транспорт. Механизм транспорта различен для разных витаминов.
В состав профилактических витаминно–минеральных комплексов наиболее часто включают в виде солей следующие макро– и микроэлементы: кальций, магний, железо, медь, йод, селен, цинк, марганец.
Как и витамины, эти минералы всасываются в основном в тонком кишечнике. Для активного транспорта во внутреннюю среду большинству из них требуются переносчики. Однако специфичность транспортного процесса не так велика, как в случае витаминов. Поэтому для минералов нередка конкуренция за общий транспортный механизм, когда присутствие в кишечнике одного минерала снижает всасывание другого. Так, в присутствии кальция и магния усвоение железа может снизиться на 50%.
Минералы могут снижать всасывание и некоторых витаминов, влияя на их растворимость или нарушая работу специфических механизмов активного транспорта. Так, ионы кальция и магния уменьшают растворимость витамина в присутствии меди, цинка или железа снижается всасывание рибофлавина.
Известны также примеры межвитаминного взаимодействия, когда один витамин инактивирует другой или нарушает его всасывание.
Так, витамин С окисляет кобаламин уже в таблетке и блокирует его всасывание при растворении таблетки в пищеварительном тракте.
Для эссенциальных микронутриентов, входящих в состав комбинированных витаминно–минеральных препаратов, известны десятки подобных негативных взаимодействий.
К сожалению, рассмотренные выше современные лекарственные формы выпуска витаминно–минеральных комплексов могут предотвратить только часть таких нежелательных взаимодействий.
Наиболее просто предотвратить нежелательный контакт компонентов в период хранения. Например, раздельное гранулирование смесей, содержащих витамины С и В12, позволяет предохранить последний от окисления.
Но если требуется учесть несколько подобных взаимодействий, то усложнение технологического процесса оказывается неприемлемым по экономическим соображениям.
Нежелательных взаимодействий микронутриентов в желудочно–кишечном тракте, когда компоненты–антагонисты имеют разные места всасывания, можно избежать, если использовать при таблетировании отличающиеся по времени растворения гранулы или делать многослойные таблетки. К сожалению, большинство микронутриентов наилучшим образом усваиваются в одной и той же зоне желудочно–кишечного тракта – в проксимальном отделе тонкого кишечника, который химус проходит за достаточно короткое время (около 3 ч).
Например, для того чтобы предотвратить снижение усвоения железа из таблетки витаминно–минерального комплекса, предлагалось помещать железо в труднорастворимое ядро таблетки, а кальций и другие двухвалентные металлы вводить в растворимый внешний слой [14]. К сожалению, метод оказался неэффективным, так как к моменту высвобождения и растворения ядра таблетка покидала оптимальное для всасывания в ЖКТ место.
Практически невозможно технологическими приемами снизить эффект негативных взаимодействий витаминно–минерального комплекса на метаболических путях организма. Для этого требуется согласованное изменение фармакокинетики компонентов [15].
Максимально эффективным методом предотвращения негативных взаимодействий между компонентами витаминно–минеральных комплексов на сегодняшний день является разделение приема микронутриентов–антагонистов по времени с интервалом в 4 часа.