Слабоумие от еды: какие продукты повышают риск болезни Альцгеймера

Болезнь Альцгеймера — это наиболее распространённая форма деменции, нейродегенеративное заболевание, впервые описанное в 1907 году немецким психиатром Алоисом Альцгеймером. Как правило, она обнаруживается у людей старше 65 лет.
Для вас всех эта тема очень важна. И вот почему:
1) Во всем мире насчитывается около 50 миллионов людей с деменцией, и ежегодно происходит почти 10 миллионов новых случаев заболевания.
2) Болезнь Альцгеймера является наиболее распространенной причиной деменции – на нее приходится 60-70% всех случаев.
3) Деменция – одна из основных причин инвалидности и зависимости среди пожилых людей во всем мире.
Развитие заболевания приводит к постепенной утрате когнитивных функций у пациентов (60-65 лет). Лица сталкиваются с потерей памяти, снижением внимания, утратой речи, пространственной ориентации, базовых навыков мышления.
СУТЬ БОЛЕЗНИ
1) Накапливается бета-амилоид – это фрагмент крупного белка, происходит скопление этих белков и отложение патологического амилоида.
-блокируется передача связи между нейронами амилоидом.
-рост количества бета-амилоида приводит к гибели нейронов.
2) Возникает воспаление
Хронические воспалительные процессы, наблюдаемые в головном мозге при БА, включают активацию микроглий и астроцитов и связаны с высвобождением свободных радикалов.
3) Тау-белок, образовавший клубочки, перестает взаимодействовать с микротрубочками и становится опасным – эти сгустки начинают повреждать нейроны. При болезни Альцгеймера тау-белки меняют форму и формируют структуры, называемые нейрофибриллярными клубками. Сплетения нарушают транспортную систему и токсичны для клеток. (микротрубочки участвуют в поддержание формы, транспорт органоидов, участвуют в делении клеток).
Гибель нейронов при всех нейродегенеративных заболеваниях осуществляется по механизму апоптоза, в основе которого лежат следующие патологические процессы: повышение концентрации аминокислот, окислительный стресс и нейровоспаление.
МЕХАНИЗМ
Многие факторы способствуют прогрессии болезни Альцгеймера, в том числе оксидативный стресс, воспаление и изменение метаболизма холестерина.
ЗАПРЕЩЕННЫЕ ПРОДУКТЫ

1) Насыщенные жиры (маргарин, свинина, сливки)
Потребление насыщенных или транснеасыщенных (гидрогенизированных) жиров повышает риск БА.
ВЛИЯНИЕ ХОЛЕСТЕРИНА НА БОЛЕЗНЬ АЛЬЦГЕЙМЕРА
Гиперхолестеринемия в среднем возрасте связана с риском развития Болезни Альцгеймера. Холестерин образует ядро бляшек амилоида.
2) Продукты с медью (Печень говяжья, кешью)

Количество меди в мозге увеличивается с возрастом и повышает выработку белка-предшественника амилоида бета. Медь увеличивает агрегацию амилоида, медь дополнительно способствует образованию реактивных форм кислорода.
Сокращение поступления меди, уменьшение потребления меди в рационе – является стратегией для регулирования образования β амилоидов.
3) Продукты с железом (Замороженная петрушка, гусиная печень)

Избыток железа способствует возникновению риска Болезни Альцгеймера. Пациенты с Болезнью Альцгеймера имеют повышенный уровень железа в областях мозга, пораженных болезнью. Избыток железа в мозге связан с образованием бета-амилоидных (Aβ) бляшек.
ВАЖНО
Поддержание нормального веса.
Cуществует связь между ожирением в среднем возрасте и слабоумием в позднем возрасте.

РАЗРЕШЕННЫЕ ПРОДУКТЫ
1) Вода
Люди с болезнью Альцгеймера забывают пить воду.
2) Витамин Е (Масло зародышей пшеницы)
 Витамин Е – антиоксидант. Защищает мозг от повреждений, вызванных свободными радикалами, воспалением.
Витамин Е – антиоксидант. Защищает мозг от повреждений, вызванных свободными радикалами, воспалением.
Высокое потребление продуктов, богатых витамином Е, снижает долгосрочный риск развития слабоумия и Болезни Альцгеймера.
3) Витамин D (Печень трески)
Люди с недостатком витамина Д больше подвержены риску развития деменции и Болезни Альцгеймера.
Витамин D снижает индуцированную амилоидами цитотоксичность и апоптоз в нейронах коры головного мозга.
Единственная бессонная ночь увеличила количество бета-амилоида в мозге

Пульсация спинномозговой жидкости в мозге человека
Nevit Dilmen / Wikimedia Commons
Исследователи в очередной раз подтвердили связь недосыпания и болезни Альцгеймера. Как утверждается в статье в Proceedings of the National Academy of Sciences, даже единственная бессонная ночь приводит к увеличению количества бета-амилоида — белка, который формирует бляшки в мозге больных.
Накопление бета-амилоида в мозге связывают с патогенезом болезни Альцгеймера — тяжелого нейродегенеративного заболевания, которое является причиной возрастной деменции в 60-70 процентах случаев. Предположительно, бета-амилоид (Aβ) накапливается в тканевой жидкости мозга в результате естественной жизнедеятельности клеток и входит в состав «метаболических отходов» мозга. За удаление отходов в центральной нервной системе отвечает так называемая глимфатическая система, которая обеспечивает обмен между тканевой жидкостью мозга и спинномозговой жидкостью. Известно, что этот обмен осуществляется за счет пульсации артерий и происходит преимущественно во время ночного сна.
Согласно описанному механизму, недостаток сна должен быть напрямую связан с накоплением в мозге «метаболических отходов», включая бета-амилоид. Экспериментально это было подтверждено на грызунах и дрозофилах с моделью болезни Альцгеймера. В нескольких работах также была показана связь между бессонницей и повышением количества бета-амилоида в тканевой жидкости мозга у здоровых людей, и связь между хроническим недосыпанием и деменцией у пожилых людей.
В новой работе сотрудники лаборатории нейровизуализации Национальных институтов здоровья США показали, что даже единственная бессонная ночь приводит к значимому увеличению концентрации бета-амилоида в мозге. Исследование включало 20 здоровых добровольцев в возрасте от 22 до 72 лет. При помощи позитронно-эмиссионной томографии у них измерили количество бета-амилоида в мозге после ночи полноценного сна (базовый уровень), а также после бессонной ночи. Измерение проводилось с использованием радиофармацевтического препарата флорбетабена, содержащего нестабильный изотоп фтора. Этот препарат связывается как с амилоидными бляшками в мозге, так и с растворимыми формами амилоида, и применяется в диагностике болезни Альцгеймера.
После бессонной ночи у 19 из 20 добровольцев количество детектируемого амилоида по сравнению с базовым уровнем значимо оказалось выше в правой подкорковой зоне мозга, включающей в себя гиппокамп. Этот отдел мозга отвечает за память и больше всего страдает при накоплении амилоидных бляшек в патогенезе болезни Альцгеймера.

Накопление изотопа в правом подкорковом кластере после бессонной ночи по сравнению с базовым уровнем. Зеленым цветом отмечен гиппокамп
Заговор с целью нейродегенерации: бета-амилоид и тау-белок
Заговор с целью нейродегенерации: бета-амилоид и тау-белок
Патогенез болезни Альцгеймера определяет тесная, но не вполне понятная связь между бета-амилоидом и тау-белком с передачей между ними токсических свойств.
Автор
Редакторы

Конкурс «Био/Мол/Текст»-2021/2022
Эта работа опубликована в номинации «Своя работа» конкурса «Био/Мол/Текст»-2021/2022.
Партнер номинации — компания Cytiva.
Генеральный партнер конкурса — международная инновационная биотехнологическая компания BIOCAD.
Генеральный партнер конкурса — компания «Диаэм»: крупнейший поставщик оборудования, реагентов и расходных материалов для биологических исследований и производств.
Бабушка без памяти от немца
Все началось в 1901 г., когда 51-летняя Августа Д., жительница Германии и супруга железнодорожного рабочего, стала вести себя странно. Внезапно фрау Д. начала сильно ревновать мужа и постепенно терять навыки ведения хозяйства. Вскоре появились нарастающие проблемы с памятью, дезориентация в пространстве и даже паранойя. Августу поместили в «Учреждение для душевнобольных и эпилептиков» во Франкфурте. Здесь ее лечением занялся врач Алоис Альцгеймер. Спустя всего 4 года Августа умерла, а ее мозг переслали переехавшему к тому времени Альцгеймеру. В этот момент кончилась история жизни Августы Д. и началась история исследования патогенеза самой значимой и известной нейродегенерации.
Алоис Альцгеймер был знаком с микроскопией ещё со времён работы над диссертацией, посвященной вырабатывающим ушную серу железам. Понимая, что он имеет дело с какой-то новой агрессивной формой «предстарческого слабоумия», врач провел тщательное патологоанатомическое исследование и микроскопию тканей мозга [1]. Он отметил гибель значительной части нейронов, а также сразу два типа патологических включений — амилоидные (или сенильные) бляшки и нейрофибриллярные клубки (рис. 1). Так свыше ста лет стали известны первые сведения о молекулярных участниках нейродегенеративной драмы [2].
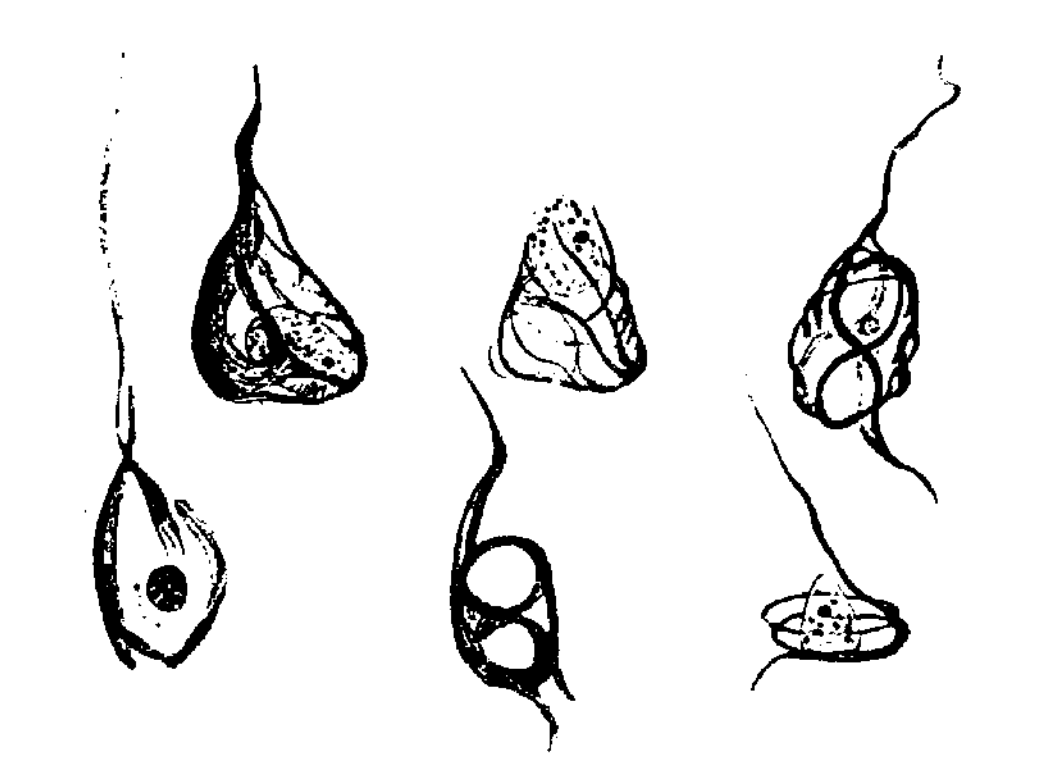
Рисунок 1. Рисунок нейрофибриллярных клубков, сделанный Алоисом Альцгеймером.
Подводя итог этой истории, стоит отметить, что если бы фрау Д. попала на прием к современному неврологу, тот скорее всего быстро поставил бы ей диагноз «деменция при болезни Альцгеймера с ранним началом». Нейропсихологические тесты подтвердили бы нарушения памяти и когнитивных способностей Августы, а нейровизуализация — потерю множества нейронов гиппокампа и коры больших полушарий. Ранее начало болезни Д. скорее всего было предопределено генетически — видимо, ей не повезло с мутациями в «проблемных» при Альцгеймере генах. Однако вряд ли сейчас ее жизнь была намного дольше или лучше. И в наши дни с момента постановки диагноза до фатального конца проходит в среднем 7–10 лет [3], [4].
Подозреваемый номер один: бета-амилоид
Итак, сенильные или амилоидные бляшки под микроскопом у Алоиса Альцгеймера в 1905 году. Слово «сенильные» понятно сходу — речь о старении (senile на английском означает «пожилой»). А вот с «амилоидом» трактовка посложнее. В основе этого слова — древнегреческое amylon («крахмал»). За амилоидами, амилоидозами и их исследованием стоит история, которая заслуживает отдельного внимания. Их веками в виде включений, подчас очень крупных, удивленные врачи извлекали из самых разных органов пациентов. Происхождение этих инородных тел и их химическая природа оставались неясными — медики просто описывали нечто «жирное на ощупь» и «похожее на воск». В середине XIX века этим вопросом занялся знаменитый Рудольф Вирхов, соавтор клеточной теории. Он заявил, что амилоиды по химической природе представляют собой крахмал — и предложил их известное нам современное название. которое совершенно неверно с точки зрения химии. В скором времени выяснилось амилоиды на самом деле представляют собой «залежи» белков, однако мы по-прежнему называем их «крахмалоподобные» [5].
Однако вернемся к Альцгеймеру и амилоиду в мозге Августы Д. Амилоид, а точнее бета-амилоид Aβ, является главным действующим лицом в истории этой нейродегенерации уже более ста лет. Aβ представляет собой полимер из примерно 40 аминокислот (как правило, 39–42) (рис. 2). Строго говоря, бета-амилоид не является белком. Имеющие менее 50 аминокислотных остатков молекулы биохимия скорее не удостоит высокого звания белка, «устройства» со сложной функцией и трехмерной структурой. Полипептид? С натяжкой, может быть. Бета-амилоидный «пептид» — более чем. Еще хороший вариант — «фрагмент белка». Корень «бета-» здесь тоже информативен: он говорит о том, что в структуре этой молекулы присутствуют бета-слои (наряду с альфа-спиралями они — базовый элемент вторичной структуры белков) [1]. И это определяет одно важное свойства Aβ — способность с легкостью образовывать «стопки»-агрегаты из «слипшихся» своей гидрофобной частью пептидов. Разделить их уже отнюдь не просто [2].

Рисунок 2. Молекулярная структура фибриллы бета-амилоида Aβ из базы данных PDB.org. Визуализация выполнена с помощью R.
Рисунок автора статьи
Откуда берется и зачем нужен такой небольшой полипептид, значительная часть которого сильно тяготеет к существованию в виде бета-тяжа? В действительности, Aβ — фрагмент более крупного белка, а именно продукт его расщепления особыми ферментами — секретазами. Этот белок имеет красноречивое название «белок-предшественник бета-амилоида» (amyloid precursor protein, APP) и обитает в мембране различных клеток, в том числе в синапсах нейронов. Здесь он имеет важное самостоятельное значение: APP необходим для нейропластичности, образования новых синапсов и в целом жизнеспособности нейронов [2]. В силу этого «зарубить амилоид на корню» вместе с APP — вряд ли хорошая идея. Работающий и сам по себе APP в какой-то момент расщепляют секретазы, которых в клетке несколько, как и соответствующих способов «нарезки» предшественника бета-амилоида. Альфа-секретаза отщепляет участок APP вблизи внешней границы мембраны клетки — это первый и универсальный этап. А вот далее этот обломок APP оказывается на распутье. Он может быть разрезан гамма-секретазой и свернуть на неамилоидогенный путь с хорошим концом, а точнее с продолжением здоровой жизни мозга (рис. 3). А может «попасть в плохие руки» бета-секретазы (aka BACE), образовать уплывающий в межклеточную среду бета-амилоид и тем запустить лавину катастрофических изменений [6].
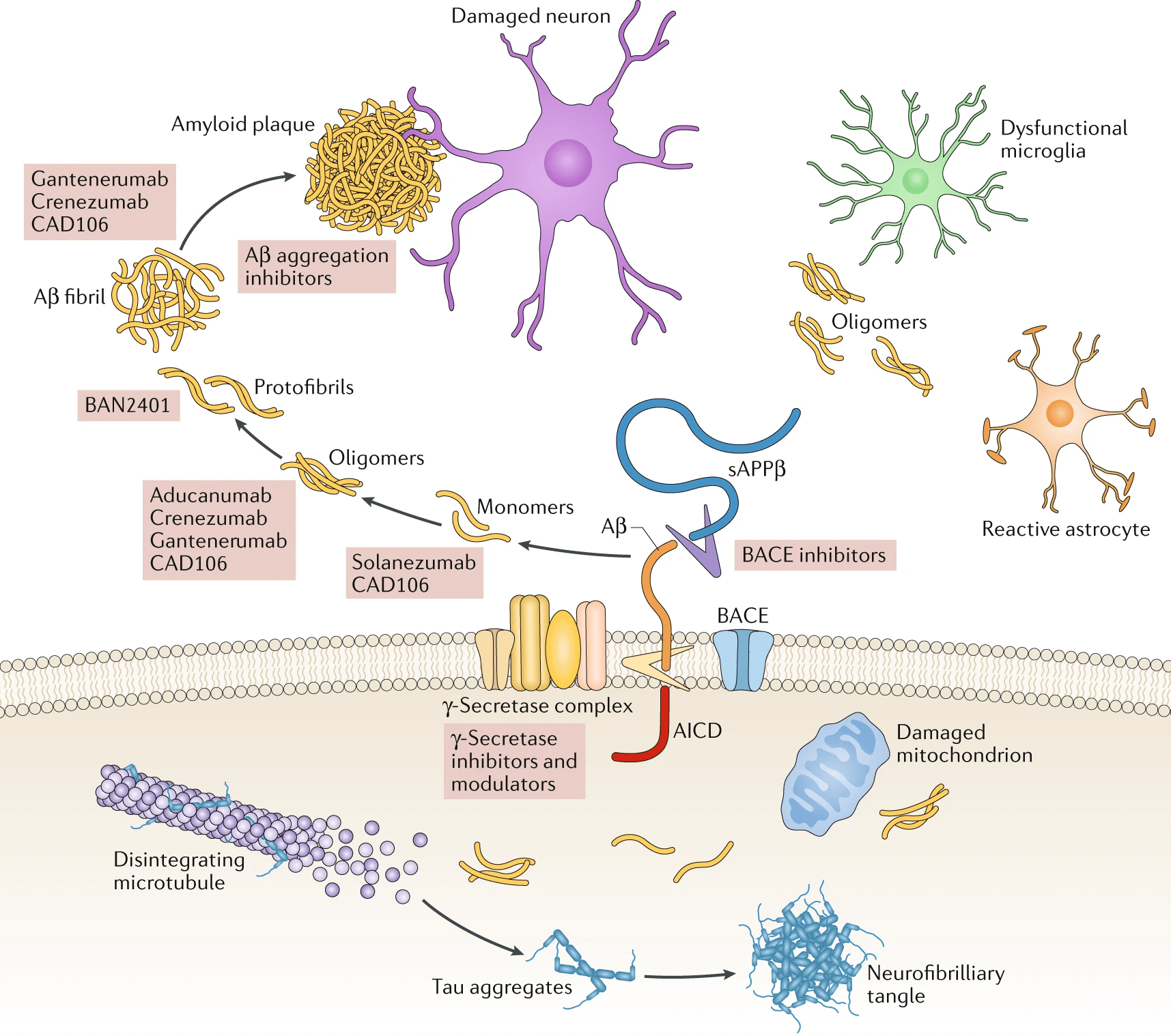
Рисунок 3. Батальное полотно, на котором изображены основные молекулярные события патогенеза болезни Альцгеймера. Сюжетная линия с бета-амилоидом располагается вне клетки («выше» клеточной мембраны), линия тау-белка — внутри.
Хотя поначалу с бета-амилоидом все не так плохо. Бывший участок APP с пропиской «в мембране, ближе к внешней части», начавший независимую жизнь растворимого бета-амилоидного пептида, сам по себе проблемой не является. Он даже стимулирует когнитивные функции мозга и образование новых нейронов [7], [8]. Но если такие молекулы образуют димер и более крупные олигомеры, это станет завязкой целого молекулярного детектива.
Дело в том, что по своей природе бета-амилоид склонен слипаться с себе подобными. На языке термодинамики это означает, что молекулам бета-амилоида энергетически выгодно соединиться — для начала в пару, а там и покучнее. А уж образовать крупные фибриллы означает для бета-амилоида и вовсе попасть в «Марианскую впадину» энергетического ландшафта и в дальнейшем уже никогда из нее не выбраться.
Для клетки эти «чисто физические» процессы не проходят даром. Димер и малые олигомеры бета-амилоида токсичны и среди прочего запускают образование активных форм кислорода [8]. А поскольку процесс распространяется, то и ситуация меняется отнюдь не в лучшую сторону. Видимо, механизм этой «эпидемии плохой конформации» (т.е. трехмерной структуры) соответствует прионной инфекции. Напомним: прионы это белки, способные передавать свои патологические свойства другим белковым молекулам. Причем не только внутри отдельно взятого организма, но и за его пределами — об этом красноречиво говорят примеры болезни Куру и губчатой энцефалопатии [9].
Любопытно отметить, что в ряде случаев патология бета-амилоида разворачивается в сосудах головного мозга. В этом случае возникают нарушения кровообращения в компании воспаления и развивается церебральная амилоидная ангиопатия.
У вас шведская или лондонская?
Склонность бета-амилоида образовывать токсичные разновидности (олигомерные, включая димеры) определяется его последовательностью. А последовательность зависит от того, какими именно секретазами был вырезан Aβ (их довольно много: альфа-, бета-, гамма-, тета-. ) и какой фрагмент APP ему «достался по наследству». Поэтому бета-амилоид может представлять собой 39, 40, 42 и т.д. аминокислотных остатков. Из них Aβ42 — форма с самым скверным характером. Он более склонен образовывать токсичные агрегаты просто в силу своих последовательности, структуры и физических свойств. Отметим: это неразрывно связанные между собой параметры пептида [10], [11].
Но это ещё не всё. Свойства бета-амилоида могут изменять и мутации, из-за которых меняются отдельные «буквы» его аминокислотной последовательности. Мутации эти, разумеется, происходят еще в кодирующем белок APP гене с неожиданным названием APP. Приходящиеся на будущий Aβ мутации меняют свойства его молекулы: либо стимулируют образование токсичных форм (патогенные мутации), либо его подавляют (протекторные мутации). Большое значение имеет то, в какую часть короткой последовательности Aβ попадет мутация. Это влияет на его взаимодействие с мембраной клетки, секретазами и точно такими же собратьями-амилоидами. В соответствии с «местом жительства» группы таких мутаций получили географические названия: лондонские, шведские, голландские и т.д. Нетрудно догадаться, что названия происходят от местности, где эти мутации были впервые описаны. А еще они соответствуют конкретным участкам бета-амилоида (рис. 4). Большинство таких «географических» мутаций не сулят ничего хорошего — их обладатель имеет повышенный шанс повстречаться на своем жизненном пути с болезнью Альцгеймера. Однако финские (совсем не как шведские) такой риск снижают [10], [12].
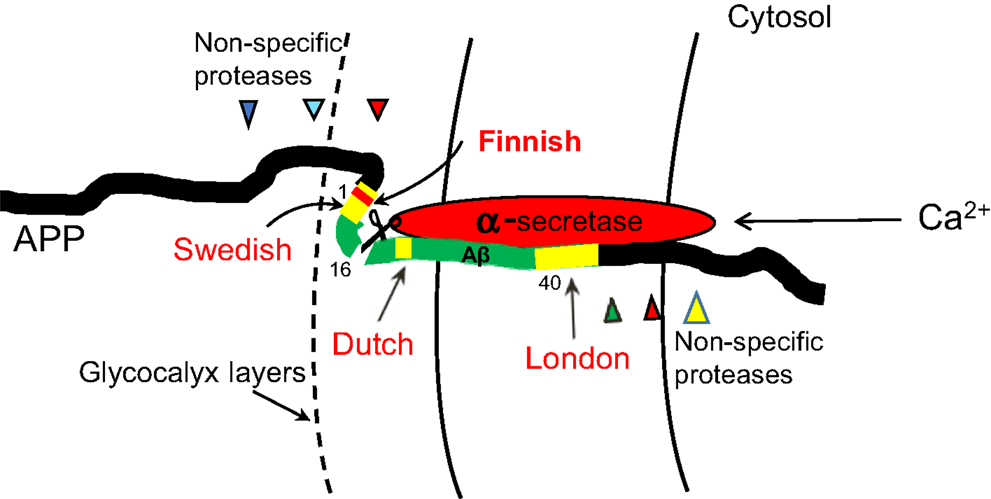
Рисунок 4. Бета-амилоид как фрагмент белка APP. Показано положение будущего Aβ относительно мембраны, сайты активности различных ферментов и локализация мутаций.
Говоря о роли мутаций в развитии нейродегенерации альцгеймеровского типа, следует заметить, что эта болезнь все же сильно зависит и от других факторов. Образ жизни, физическая активность, уровень интеллектуальной деятельности, питание и т.д. могут значительно изменить риск ее развития. Этого нельзя сказать о неотвратимых генетических поломках при «сугубо наследственных» болезнях вроде нейродегенерации с накоплением железа в мозге (ННЖМ) или болезни Баттена. Если есть мутация, почти наверняка будет и болезнь, а без мутации ее можно не опасаться. Не менее важно, что «замешанные» в Альцгеймере мутации не ограничиваются сугубо геном APP. Очень велико также значение генов, кодирующих различные секретазы (известны как пресенелины: PSEN1 и PSEN2), аполипопротеин E (APOE)… список на самом деле довольно длинный [4], [10].
А вот гена MAPT — инструкции по изготовлению тау-белка — среди них нет. Автора в свое время это удивило. Оказывается, участие тау-белка в альцгеймеровской нейродегенерации скорее зависит не от его последовательности, а от разнообразия изоформ — продуктов альтернативного сплайсинга этого гена [10], [13].
Подозреваемый номер два: тау-белок
Прежде чем начинать вторую сюжетную линию болезни Альцгеймера, стоит отойти на шаг назад. Зачем в ней вообще понадобился второй главный герой? Казалось бы, все дело в бета-амилоиде и его прямом либо косвенном разрушительном действии. Известны конкретные мутации в гене APP и их трагические последствия. Aβ — зло, его отсутствие — благо, и стоит избавиться от него, как проблема будет решена.
Однако в последние десятилетия поступают данные о том, что не все с ним так просто. Бета-амилоид и, в частности, сенильные бляшки обнаружили и в мозге здоровых людей, причем не только пожилых. Свободный бета-амилоид считают необходимым для когнитивных функций и даже иммунитета. А заболевшие могут иметь незначительные отложения Aβ на фоне тяжелой симптоматики или наоборот. Иными словами, нет корреляции между количеством Aβ в мозге и тяжестью нейродегенерации. В то же время для тау-белка и образованных им нейрофибриллярных клубков эта корреляция четко прослеживается [2], [4], [14].

Рисунок 5. Структура олигомеров тау-белка (τ-белка) из базы данных PDB.org. Визуализация выполнена с помощью R.
Рисунок автора статьи
Что скрывается за таинственным названием «тау-белок»? Ассоциированный с микротрубочками белок тау или microtubule-associated protein tau (MAPT) (рис. 5). Наряду с несколькими другими, впрочем, менее примечательными (видимо, они даже не стоили того, чтобы выделить им собственную греческую букву) MAPT стабилизирует «внутриклеточную арматуру» — микротрубочки нейронов. От других MAP-ов тау-белок отличает место работы — преимущественно мозг: нейроны и, реже, клетки глии. Любопытно, что в мозге он все же не единственный «микротрубочковый стабилизатор» — нокаутированные по гену MAPT мыши вполне жизнеспособны. А его главная функция как раз в этом — стабилизировать микротрубочки и не давать им диссоциировать. А постабилизировать в нейронах есть чего. Вдоль аксонов нейронов протянуты как раз микротрубочки, поддерживающие их форму и необходимые для аксонального транспорта [15].
В качестве посттрансляционной модификации тау-белок предпочитает фосфориловаться. Фосфорилирование (т.е. «навешивание» фосфатов) происходит по определённым положениям MAPT. Оно значительно меняет способность тау-белка выполнять свою главную задачу — связывать микротрубочки и удерживать их вместе. По тем или иным причинам тау-белок может оказаться фосфорилирован слишком сильно и не там, где следует (это уже так называемый гиперфосфорилированный тау). В этом случае он выпускает свой любимый лиганд (микротрубочки), и те начинают разбираться на части.
Сам тау-белок является внутренне неупорядоченным (intrinsically disordered) и обретает определенную структуру только в связанном с лигандом виде. Он ведёт себя подобно Aβ и образует патологические агрегаты — парные спиральные филаменты (paired helical filaments, PHF) и сложенные из них более крупные нейрофибриллярные клубки (neurofibrillary tangles). А последние, напомним, рассмотрел в микроскоп ещё Альцгеймер [1], [2].
Что заставляет тау-белок вести себя таким разрушительным образом? Мы уже упоминали, что это не мутации в соответствующем гене MAPT. Нельзя сказать, что это такой исключительный ген без мутаций: среди таупатий (группы связанных с патологией белка заболеваний) есть и генетические, вызванные именно мутациями в MAPT. Хороший пример — лобно-височная деменция и паркинсонизм, связанные с 17-й хромосомой (FTDP-17), прогрессирующий супрануклеарный паралич и кортикобазальная дегенерация.
Однако в случае связанной с тау-белком сюжетной линии болезни Альцгеймера дело не в мутациях. Во-первых, его патологические состояния связаны с изоформами — продуктами его сплайсинга. Всего таких форм известно 6, и среди них наибольшую токсичность имеет 4R-тау белок. Четверка в этом случае означает, что в аминокислотную последовательность попали четыре копии области тандемных повторов, а не три [2], [13].
Другим важным фактором «плохого поведения» тау белка является дурное влияние бета-амилоида. Все больше сторонников приобретает концепция о том, что Aβ служит своего рода зачинщиком разрушительных процессов, которые по большей части берет на себя уже тау-белок. Считается, что токсичные олигомерные формы с прионными свойствами бета-амилоида порождают токсичные олигомерные формы с прионными свойствами уже белка тау. И болезнь Альцгеймера оказывается захватывающей и запутанной «историей двух прионов» [2], [17].
Каким образом это пагубное влияние разворачивается в клетке. Или где-то ещё?
Здесь хотелось бы отметить важный момент, который нередко игнорируют — может быть, считают самоочевидным. Как правило, Aβ и тау имеют довольно мало шансов напрямую повстречаться в мозге. Бета-амилоид начинает свою независимую жизнь, когда гамма-секретаза вырезает его из APP у внешней части мембраны. После этого Aβ отправляется в свободное плавание по внеклеточной среде. Тем временем действие с участием тау-белка разворачивается внутри нейронов (рис. 3). Каким же образом они взаимодействуют, передавая, как эстафетную палочку, прионные свойства и сообща разрушают мозг? Вопрос более чем открытый.
Естественно думать, что их взаимодействие чем-то опосредованно. И самое очевидное — различные сигнальные пути клетки, благодаря которым она способна управлять своим состоянием. Действительно, известно, что патологические формы бета-амилоида запускают процессы воспаления, а также активируют протеинкиназы — ферменты, фосфорилирующие белки. В этом отношении особенно важна протеинкиназа под названием GSK. О дальнейшем догадаться нетрудно: протеинкиназы делают тау гиперфосфорилированным и тем запускают усиливающую сeбя патологию. Однако детали этого перехода «амилоид/тау» не вполне ясны. Интересно, что «переброситься» от одного к другому патология, по-видимому, все же может и напрямую. Известно, что физическое взаимодействие бета-амилоида и тау-белка может вызывать прямую передачу «плохой конформации». Повторимся: как именно происходит этот, значимый для всего патогенеза процесс, пока не ясно [2].
Заговор и прочие подельники
Помимо бета-амилоида и тау-белка в альцгеймеровской нейродегенерации есть и другие участники. Более того, весь стоящий за патогенезом криминальный синдикат повязан воедино. Наука давно ушла от линейности и однонаправленности процессов, оставив ее на классическом этапе своего развития. И это полезно помнить, рассматривая вносящие свой вклад в нейродегенерацию Aβ, тау и других немаловажных игроков [18].
Ацетилхолин и базальные ядра
Когда врачи приступили к поиску лекарств болезни Альцгеймера, они обратили внимание на дефицит в мозге заболевших такого важного нейромедиатора, как ацетилхолин. Именно он стоит за многими симптомами вроде нарушений памяти, психики и т.д. Однако это, безусловно, вторичное проявление болезни, которое просто «визуализирует» гибель нейронов в одном из ключевых локаций мозга — холинергических клеток базального переднего мозга [19].
Окислительный стресс
Есть мнение, что если вы не знаете в чем причина того или иного заболевания, валите все на окислительный стресс и активные формы кислорода (АФК). В этом есть доля правды. В случае болезни Альцгеймера АФК, в частности, напрямую и очень эффективно производит бета-амилоид. Важен окислительный стресс и как «отягчающее обстоятельство», усиливающее другие негативные явления вроде хронического воспаления.
С окислительным стрессом, как это водится, под руку ходит нарушение состояния митохондрий. Действительно, ряд работ указывают на особую их роль в патогенезе болезни Альцгеймера.

Рисунок 6. Модели взаимодействия бета-амилоида с мембраной. Слева — образованная пептидом пора.
Ну а где окислительный стресс и митохондрии, там ещё и изменения в мембранах. Ненасыщенные жирнокислотные хвосты фосфолипидов, которые они стыдливо прячут в силу гидрофобности, особенно чувствительны к окислительному стрессу. Помимо этого, есть и ряд специфических мембранных проблем при болезни Альцгеймера. Так, гидрофобный и, вероятно, помнящий свое «мембранное прошлое» амилоид может встраивается в нее снова и даже, видимо, образовывать поры (рис. 7). Если этот сценарий справедлив, через эти поры начинают неизбирательно проходить разные не предназначенные для этого вещи. Так или иначе, Aβ меняет состояние липидного бислоя и отнюдь не в лучшую сторону. Ну а в мембранах, как известно, несут свою службу ионные каналы и различные клеточные рецепторы [2], [20].
Клеточные рецепторы и кальциевая сигнализация
Кажется, в этот момент мы собрали вокруг патогенеза болезни Альцгеймера все излюбленные проблемы биофизики. Разве что без фотобиологии — по крайней мере, пока. Так вот, изменения состояния мембраны сказывается на различных проводящих, узнающих и усиливающих сигнал рецепторах клеточной мембраны. Как напрямую, так и за счет изменения ее проводимости. Среди таких путей сигнализации особенно важна опосредованная кальцием. Все же нейрон — клетка возбудимая и без потенциала действия не обходится. Есть данные о прямом вмешательстве бета-амилоида в этот тонкий механизм [21].
В этот список можно включить и хроническое воспаление, и аутофагию, и много еще чего — все упомянутое лишь иллюстрирует разнообразие механизмов и переплетенные связи между. Разобраться в них чрезвычайно важно — чтобы распутать этот клубок, найдя его тонкие и слабые места.
Забыть Альцгеймера
Все сказанное, безусловно, должно стать основой для создания, выражаясь языком английской кальки, «механистического лечения». То есть такого, которое действительно работает. Безусловно, должно и, безусловно, до сих пор не может.
Болезнь Альцгеймера изучают более ста лет. Эта тематика привлекла огромные усилия и колоссальные денежные вливания. Но лекарства все нет, а заболевших в мировом масштабе 30 миллионов. Это стареющий мир… все большее число людей «доживают до своего Альцгеймера». Поэтому положение вещей станет только хуже. Прогнозы ВОЗ утверждают: хуже впятеро. Ожидается, что к 2050 году лиц с нейродегенерацией Альцгеймеровского типа станет 150 млн [5], [22].
Конечно, сейчас есть несколько одобренных (в том числе FDA) препаратов. Это 3 ингибитора холинэстеразы (донепезил, ривастигмин, галантамин) и регулятор глутаматных рецепторов мемантин. Все эти препараты способны лишь немного смягчить проявления болезни Альцгеймера. И этот список не пополнялся с 2003 года, когда в него последним вошёл мемантин [21].
Некогда большие надежды связывали с иммунотерапией болезни Альцгеймера. Казалось, решение найдено. Виновник — Aβ — можно устранить, натравив на него иммунную систему, с помощью нацеленных на него антител — «поцелуев смерти». Однако эти ожидания оказались во многом обмануты. Не только болезнь Альцгеймера не удалось излечить с помощью иммунотерапии: врачи ещё и столкнулись с тяжелыми побочными эффектами. В чем причина этой неудачи? Видимо, дело опять-таки в том, что ее патогенез не ограничивается амилоидом единым, да и сам амилоид оказался слишком полиморфным, т.е. существующим в виде различных по размеру и поведению форм [23]. Впрочем, исследования продолжаются, а препарат Aducanumab (моноклональное антитело, как нетрудно узнать по соответствующему трехбуквенному окончанию -mab) в клинических испытаниях смог улучшить состояние больных. Однако Aducanumab, видимо, не ограничивается действием исключительно на бета-амилоид [24].
Рисунок 7. У нейрона много врагов и ему нужна помощь.
Коллаж автора статьи
Однако вооружаясь «геномными инструментами», нужно, наконец, понять, что же в хитросплетении болезни Альцгеймера имеет решающее значение. И бить по умело выбранной цели.