Клинические исследования. Фазы, виды, условия.
Департамент Клинических Исследований (Clinical Trial Unit) клинико-диагностического центра «Евромедсервис» рад приветствовать Вас на нашем сайте и рассказать о Клинических Исследованиях.
Клинические исследования лекарственного средства являются необходимым этапом разработки нового препарата, или расширения показаний для применения лекарственного средства, уже известного врачам.
· На какие этапы делятся клинические исследования?
Выделяют 4 фазы (этапа) клинических исследований:
Фаза I – Проверка механизма действия (длительность: от нескольких месяцев до 1 года):
в исследование включается небольшая группа здоровых добровольцев. Участникам увеличивают дозы препарата, начиная с минимальной и до максимально допустимой. После каждого приема отслеживается состояние пациента. Выясняется предпочтительный вариант применения и уровень дозировки.
группа пациентов по заболеванию увеличивается, в течение этой фазы определяется краткосрочная безопасность лекарственного препарата (фаза IIa), эффективность лекарственного препарата и его дозировка (фаза IIb), насколько новый препарат эффективнее по сравнению с плацебо или уже существующим лечением.
Фаза III – Подтверждающие исследования (длительность: от года до нескольких лет)
на данном этапе происходит полномасштабная оценка показателя риск/польза. самый длительный этап клинических исследований с самым большим количеством включаемых пациентов (тысячи пациентов в разных странах), которая проводится в разных исследовательских центрах в различных странах. Цель: подтверждение эффективности и безопасности исследуемого вещества на больших выборках. Министерство Здравоохранения РФ принимает решение о выдаче регистрационного удостоверения лекарственного препарата, позволяющего вывести его на рынок.
Фаза IV – проводится после получения регистрационного удостоверения лекарственного препарата с целью получения расширенных данных по его безопасности, а также социо-экономических данных.
Что понимают в клинических исследований под термином «Дизайн»?
Дизайн – это способ проведения научного исследования в клинике, т.е. его организация или архитектура.
По дизайну клинические исследования могут быть:
e) сравнительными – применение нового исследуемого лекарственного препарата сравнивается с уже существующими методами лечения или лекарственными препаратами.
· Зачем участвовать в клинических исследованиях?
Ни один новый препарат и новый способ лечения в мире не может быть разрешен к широкой продаже без проведения необходимых клинических исследований. Участники клинических исследований получают бесплатный доступ к новым исследуемым видам лечения до их широкого распространения.
· Каковы риски при участии в клиническом исследовании?
Такие же, как и при лечении заболевания в условиях обычной клинической практики: риск побочных эффектов и риск осложнений от медицинских процедур и манипуляций.
· Нужно ли платить за участие в КИ?
Участие в исследовании, включая все определенные Протоколом процедуры и терапию, являются бесплатными для участника исследования.
· Какие права и обязанности есть у участника клинического исследования?
Обязательно в письменном виде пациенту предоставляется информация о цели КИ, о том, как оно будет проводиться, какое лекарство будет применяться, и т.д.
Всё изложенное отражено в особом документе — информированном согласии. Информация, позволяющая идентифицировать участника исследования кодируется и сохраняется в тайне на протяжении всего участия в исследовании и после завершения участия в нем.
С момента подписания формы информированного согласия участник исследования застрахован от причинения вреда жизни и здоровью. Пострадавшим в результате участия в исследовании гарантированы соответствующие компенсация и лечение.
Участник клинического исследования берет на себя обязанность соблюдать все требования врача и Протокола исследования (графики процедур и визитов в клинику, график приема исследуемого лекарственного препарата и т.д.) и сообщать врачу, когда эти требования были нарушены по какой-либо причине.
Участник может покинуть клиническое исследование в любое время, независимо от причины.
Виды клинических исследований лекарственных средств
Клинические исследования лекарственного средства являются необходимым этапом разработки любого нового препарата, или расширения показаний для применения лекарственного средства, уже известного врачам. На начальных этапах разработки лекарственных средств проводятся химические, физические, биологические, микробиологические, фармакологические, токсикологические и другие исследования на тканях (in vitro) или на лабораторных животных. Это так называемые доклинические исследования, целью которых является получение научными методами оценок и доказательств эффективности и безопасности лекарственных средств. Однако эти исследования не могут дать достоверной информации о том, как изучаемые препараты будут действовать у человека, так как организм лабораторных животных отличается от человеческого и по фармакокинетическим характеристикам и по реакции органов и систем на лекарства. Поэтому необходимо проведение клинических испытаний лекарственных средств у человека.
Итак, что такое клиническое исследование (испытание) лекарственного средства? Это системное изучение лекарственного препарата посредством применения его у человека (пациента или здорового добровольца) с целью оценки его безопасности и/или эффективности, а также выявления и/или подтверждения его клинических, фармакологических, фармакодинамических свойств, оценки всасывания, распределения, метаболизма, выведения и/или взаимодействия с другими лекарственными средствами. Решение о начале клинического исследования принимает Спонсор/Заказчик, который несет ответственность за организацию, контроль и/или финансирование исследования. Ответственность за практическое проведение исследования возложена на Исследователя (лицо или группу лиц). Как правило, спонсором являются фармацевтические компании – разработчики лекарственных средств, однако в роли спонсора может выступать и исследователь, если исследование начато по его инициативе и он несет полную ответственность за его проведение.
Клинические исследования должны проводиться в соответствии с основополагающими этическими принципами Хельсинкской Декларации, Правилами GCP (Good Clinical Practice, Надлежащая Клиническая Практика) и действующими нормативными требованиями. До начала клинического исследования должна быть проведена оценка соотношения предвидимого риска с ожидаемой пользой для испытуемого и общества. Во главу угла ставится принцип приоритета прав, безопасности и здоровья испытуемого над интересами науки и общества. Испытуемый может быть включен в исследование только на основании добровольного информированного согласия (ИС), полученного после детального ознакомления с материалами исследования.
Клиническое исследование должно быть научно обосновано, подробно и ясно описано в протоколе исследования. Оценка соотношения рисков и пользы, а также рассмотрение и одобрение протокола исследования и другой документации, связанной с проведением клинических исследований, входят в обязанности Экспертного Совета Организации / Независимого Этического Комитета (ЭСО / НЭК). После получения одобрения от ЭСО/НЭК можно приступать к проведению клинического исследования.
Виды клинических исследований
Пилотное исследование предназначено для получения предварительных данных, важных для планирования дальнейших этапов исследования (определение возможности проведения исследования у большего числа испытуемых, размера выборки в будущем исследовании, необходимой мощности исследования и т.д.).
Рандомизированное клиническое исследование, в котором пациенты распределяются по группам лечения случайным образом (процедура рандомизации) и имеют одинаковую возможность получить исследуемый или контрольный препарат (препарат сравнения или плацебо). В нерандомизированном исследовании процедура рандомизации не проводится.
Контролируемое (иногда используется синоним «сравнительное») клиническое исследование, в котором исследуемое лекарственное средство, эффективность и безопасность которого до конца еще не изучены, сравнивают с препаратом, эффективность и безопасность которого хорошо известны (препарат сравнения). Это может быть плацебо, стандартная терапия или отсутствие лечения вообще. В неконтролируемом (несравнительном) исследовании группа контроля / сравнения (группа испытуемых, принимающих препарат сравнения) не используется. В более широком смысле под контролируемым исследованием имеется в виду всякое исследование, в котором контролируются (по возможности минимизируются или исключаются) потенциальные источники систематических ошибок (т. е. оно проводится в строгом соответствии с протоколом, мониторируется и т.д.).
При проведении параллельных исследований испытуемые в различных группах получают либо только изучаемое лекарственное средство, либо только препарат сравнения / плацебо. В перекрестных исследованиях каждый пациент получает оба сравниваемых препарата, как правило, в случайной последовательности.
Исследование может быть открытым, когда все участники исследования знают, какой препарат получает пациент, и слепым (замаскированным), когда одна (простое слепое исследование) или несколько сторон, принимающих участие в исследовании (двойное слепое, тройное слепое или полное слепое исследование) держатся в неведении относительно распределения пациентов по группам лечения.
Проспективное исследование проводится с делением участников на группы, которые будут или не будут получать исследуемое лекарственное средство, до того, как наступили исходы. В отличие от него, в ретроспективном (историческом) исследовании изучаются исходы проведенных ранее клинических исследований, т.е. исходы наступают до того, как начато исследование.
В зависимости от количества исследовательских центров, в которых проводится исследование в соответствии с единым протоколом, исследования бывают одноцентровыми и многоцентровыми. Если исследование проводится в нескольких странах, его называют международным.
В параллельном исследовании сравниваются две или более группы испытуемых, одна или более из которых получают исследуемый препарат, а одна группа является контрольной. В некоторых параллельных исследованиях сравнивают различные виды лечения, без включения контрольной группы. (Такой дизайн называют дизайном независимых групп).
Когортное исследование – это обсервационное исследование, в котором выделенную группу людей (когорту) наблюдают в течение некоторого времени. Исходы у испытуемых в разных подгруппах данной когорты, тех кто подвергался или не подвергался (или подвергался в разной степени) лечению исследуемым препаратом сравниваются. В проспективном когортном исследовании когорты составляют в настоящем и наблюдают их в будущем. В ретроспективном (или историческом) когортном исследовании когорту подбирают по архивным записям и прослеживают их исходы с того момента по настоящее время.
В исследовании случай-контроль (синоним: исследование сходных случаев) сравнивают людей с определенным заболеванием или исходами («случай») с людьми из этой же популяции, не страдающими данным заболеванием, или у которых не наблюдался данный исход («контроль»), с целью выявления связи между исходом и предшествующему воздействию определенных риск-факторов. В исследовании серии случаев наблюдают несколько индивидуумов, обычно получающих одинаковое лечение, без использования контрольной группы. В описании случая (синонимы: случай из практики, история заболевания, описание единичного случая) ведется исследование лечения и исхода у одного человека.
В настоящее время предпочтение отдается такому дизайну клинического исследования лекарственных средств, при котором обеспечивается получение наиболее достоверных данных, к примеру, при проведении проспективных контролируемых сравнительных рандомизированных и, желательно, двойных слепых исследований.
В последнее время роль клинических исследований лекарственных средств возросла в связи с внедрением в практическое здравоохранение принципов доказательной медицины. И главным среди них является принятие конкретных клинических решений для лечения пациента на основе строго доказанных научных данных, которые могут быть получены в ходе хорошо спланированных, контролируемых клинических исследований.
Прагматические клинические исследования
Полный текст:
Аннотация
Прагматические исследования позволяют объединить преимущества наблюдательного исследования в реальной клинической практике с научной строгостью рандомизированного клинического исследования (РКИ) и тем самым дать более эффективные ответы на вопросы реальной клинической практики.
Цель. Оценка различий в проведении классических РКИ и прагматических клинических исследований (ПКИ), а также анализ особенностей, касающихся их проведения на разных этапах.
Методология. Проведён анализ ряда публикаций в период с 1999 по 2017 гг. на предмет выявления данных, посвящённых ПКИ.
Результаты. В проведении классических РКИ и ПКИ имеются существенные различия. Прежде всего, в ПКИ используют более гибкие критерии включения и отличается подход к выбору центра исследования. Также процедура получения информированного согласия имеет существенные отличия от таковой при классическом РКИ; предложены альтернативные варианты, однако единый подход пока не выработан. При проведении ПКИ требуются минимальные вмешательства монитора в рутинную медицинскую практику, что, однако, может привести к нарушению сбора отчётности. Вариантом решения может стать удалённый сбор данных.
Выводы. ПКИ представляют собой огромный потенциал для исследования эффективности лекарственных препаратов в условиях реальной клинической практики. Однако, несмотря на значительное увеличение количества таких исследований, всё ещё существует достаточное количество моментов, требующих урегулирования.
Ключевые слова
Для цитирования:
Шевченко О.Р., Колбин А.С. Прагматические клинические исследования. Качественная Клиническая Практика. 2020;(3):52-60. https://doi.org/10.37489/2588-0519-2020-3-52-60
For citation:
Shevchenko O.R., Kolbin A.S. Pragmatic clinical trials. Kachestvennaya Klinicheskaya Praktika = Good Clinical Practice. 2020;(3):52-60. (In Russ.) https://doi.org/10.37489/2588-0519-2020-3-52-60
Актуальность
Рандомизированные клинические исследования (РКИ) проводят с середины прошлого века, и их роль в современной медицине сложно переоценить. Основываясь на данных классического РКИ, под которым подразумевается двойное слепое рандомизированное плацебо-контролируемое исследование, можно судить о действенности (англ. efficacy) лекарственного препарата (ЛП). Данный вид исследований чаще всего используют при проведении предрегистраци-онных интервенционных клинических исследований [1]. Однако, даже если данные, полученные в ходе качественно проведённого РКИ, валидны, это не гарантирует получения такого же результата у пациентов, не соответствующих субъектам, подобранным по строгим критериям для участия в исследовании. В связи с этим, существует всё больше запросов на получение информации об эффективности ЛП в условиях реальной клинической практики (англ. Real-World Data, RWD).
Одно из определений RWD — «данные, относящиеся к системе здравоохранения, которые собирают из различных источников вне рамок предре-гистрационных рандомизированных клинических исследований» [2].
После анализа таких данных получают доказательства реального мира (англ. Real-World Evidence, RWE), с получением данных об эффективности (англ. effectiveness) [3].
В медицинском сообществе сложилось мнение, что основным источником данных RWD являются наблюдательные исследования [2, 4]. Как классическое РКИ, так и наблюдательные исследования имеют ограничения в предоставлении данных о сравнительной эффективности ЛП.
Недавнее метаэпидемиологическое исследование показало, что после проведённого РКИ можно получить совершенно разные ответы на одни и те же клинические вопросы в результате рутинного сбора данных о состоянии здоровья, что, несмотря на трудности в устранении статистических погрешностей, может привести к переоценке эффекта лечения [5]. Также при проведении РКИ минусом является исследование ограниченных групп населения в строго подобранных и контролируемых условиях, оптимизированных для демонстрации действия ЛП [6].
В случае с наблюдательными исследованиями нельзя исключить наличие систематических ошибок ( англ. bias), особенно, касающихся несопоставимости между группами пациентов [5].
Необходимо отметить, что в настоящий момент огромное количество данных о состоянии здоровья пациентов хранится на компьютерах, мобильных устройствах и других гаджетах. Эти данные могли бы быть полезны, чтобы лучше спланировать проведение клинических исследований.
Многими экспертами в качестве дополнительного источника данных о RWD предлагается рассматривать прагматические клинические исследования (ПКИ, англ. pragmatic clinical trials), которые позволяют объединить преимущества наблюдательного исследования в реальной клинической практике с научной строгостью рандомизированного клинического исследования, и тем самым более точно ответить на вопросы реальной клинической практики.
Цель: анализ особенностей проведения прагматических клинических исследования.
Методология
Был проведён систематический анализ данных литературы в период с 1967 по 2019 гг. на предмет выявления информации, посвящённой ПКИ. В ходе проведения поискового анализа использовали следующие критерии включения и исключения.
Критерии включения: в анализ вошли публикации, в которых описывали исследования по эффективности, сравнительной эффективности (наблюдательные исследования, прагматические клинические исследования).
Критерии исключения: в анализ не вошли клинические исследования, посвящённые обсуждению действенности и сравнительной действенности.
Ключевые слова поиска: клинические исследования; прагматические клинические исследования; реальная клиническая практика; исследования сравнительной эффективности.
Результаты
История. Впервые прагматические клинические исследования были предложены и описаны Schwartz D. и Lellouch J. в 1967 году [7]. В этой статье авторы представили два сценария испытаний по борьбе с раком, в которых исследовали лекарственный препарат, повышающий чувствительность к радиотерапии. Предположив, что ЛП следует вводить на протяжении 30 дней до радиотерапии, группы сравнения можно было сформировать двумя способами, которые представлены на рис. 1.
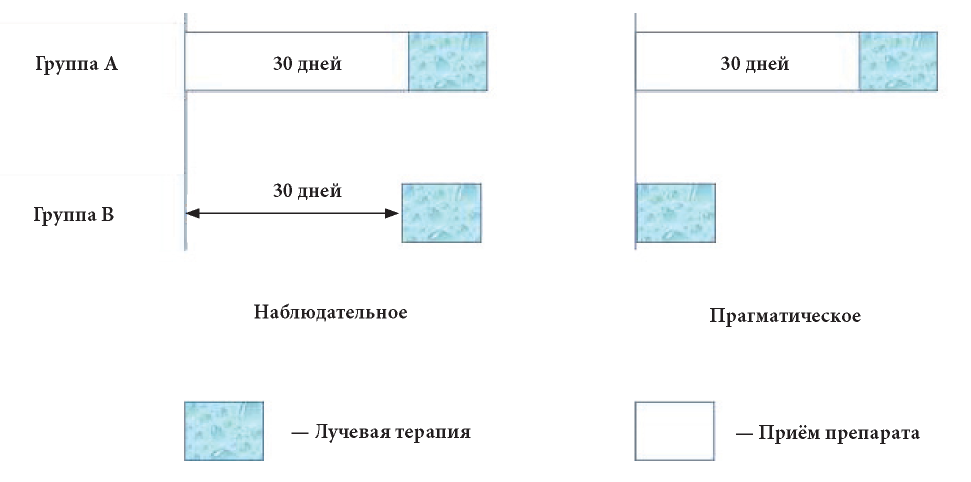
Рис. 1. Наблюдательный и прагматический подход в исследованиях [4]
Как видно из данных, представленных на рис. 1, чтобы при наблюдательном исследовании лучевая терапия проводилась в одно и то же время в каждой группе, в группе сравнения (Группа Б) лучевой терапии предшествует «пустой» период в 30 дней (приём препарата и другие вмешательства не осуществляются). Во втором случае (прагматическое исследование) лучевая терапия Группы Б могла быть начата одновременно с приёмом лекарственного препарата Группой А. Ни один из этих вариантов процедур нельзя было назвать «лучшим» по отношению к другому. Первый (наблюдательное исследование) позволял сравнивать две группы, которые похожи с точки зрения лучевой терапии и которые отличались только наличием или отсутствием ЛП. Была проведена оценка сенсибилизирующего действия лекарственного препарата. Вторая процедура (прагматическое исследование) позволила сравнить две процедуры в тех условиях, в которых они наиболее вероятно будут применяться на практике. Таким образом, это два абсолютно разных подхода к исследованию ЛП.
CONSORT. По мере роста количества клинических исследований также возросла необходимость обеспечения чёткого представления результатов. Для достижения этой цели были созданы Единые стандарты представления результатов исследований (англ. Consolidated Standards of Reporting Trials, CONSORT) и выпущен контрольный лист CONSORT, состоящий из 22 пунктов. В 2008 году было выпущено расширение заявления CONSORT для прагматических клинических исследований, которое не изменило основные пункты контрольного листа, однако внесло дополнения в 8 из них [8]. Расширения затронули такие пункты, как: исходные данные, участники исследования, вмешательства, результаты, размер выборки, ослепление, набор участников и обобщение результатов. Для каждого из восьми пунктов представлены стандартный текст CONSORT и дополнительные указания, пример хорошей отчётности по данному пункту и объяснение проблем. Важно отметить, что эти предложения следует рассматривать как дополнение к общим указаниям в основном пояснительном документе CONSORT и, при необходимости, к другим указаниям CONSORT. Эти расширения представлены вместе с иллюстративными примерами отчётности и объяснением каждого расширения.
Колесо PRECIS-2. При планировании проведения исследования сравнительной эффективности существующей методологии или лекарственного препарата следует сделать выбор в пользу прагматического или наблюдательного подхода, поскольку они представляют собой две противоположные стороны одного континуума. С этой целью в 2005—2008 гг. команда из 25 международных экспертов и методистов разработала инструмент PRECIS ( англ. Pr agmatic- E xplanatory C ontinuum I ndicator S ummary) [9]. Далее первоначальная версия была усовершенствована, чтобы на этапе разработки исследований выбрать наиболее верный подход в соответствии с планируемой целью. Так, разработчиками было представлено «колесо» PRECIS-2 (рис. 2).
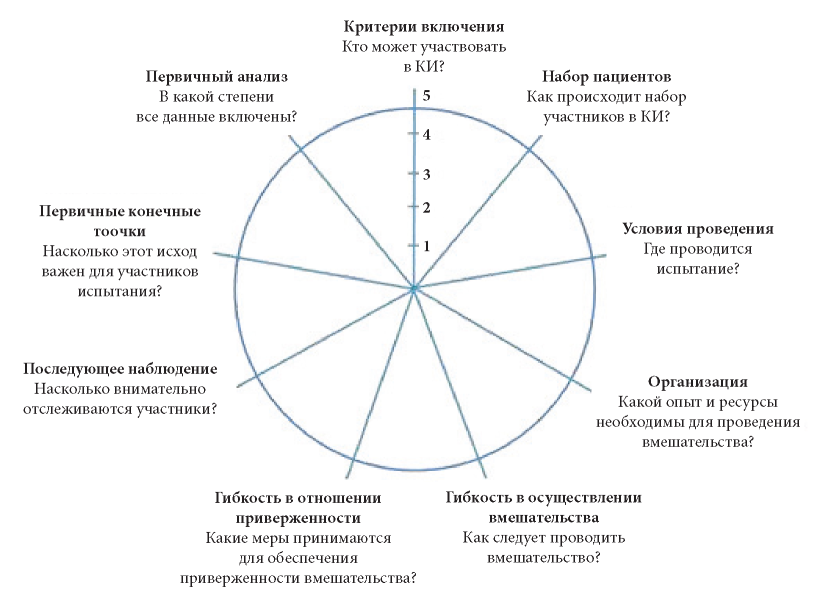
Рис. 2. Модель (колесо) PRECIS-2 [10]
Как видно из данных, представленных на рис. 2, представлен инструмент, который поможет выбрать наиболее соответствующий целям исследования подход. Модель (колесо) PRECIS-2 имеет 9 пунктов, каждый из которых оценивается по пятибалльной шкале, где 1 — «очень наблюдательные», идеальные условия, а 5 — «очень прагматичные условия», соответствующие реальной клинической практике. Целью «очень прагматичного» подхода в данном случае считается максимальное применение вмешательства для обычной помощи в реальной клинической практике и его эффективность, в то время как «очень наблюдательный» подход демонстрирует эффект вмешательства с помощью ожидаемого механизма, однако вопросу о том, будет ли этот результат достигнут в реальных условиях, уделяется минимальное внимание. К примеру, при выборе центра проведения исследования обычные стационары, осуществляющие рутинную медицинскую деятельность, будут соответствовать 5 баллам (прагматичный подход), в то время как специализированные для проведения исследований центры наберут 1 балл. Таким образом, используя это колесо решений, исследователям и клиницистам будет легче понять, соответствуют ли проектные решения их предполагаемому назначению.
Отличия в проведении ПКИ от классического РКИ. В табл. 1 суммированы и представлены основные отличия классического РКИ от прагматического.
Отличия классического рандомизированного клинического исследования от прагматического клинического исследования
Классическое рандомизированное клиническое исследование
Прагматическое клиническое исследование
По строгим критериям
В условиях реальной клинической практики
в проведении исследований
стандартная утверждённая форма
Возможно использовать упрощённую форму
о нежелательных реакциях
Стандартная форма отчётности
Различные источники, форма отчётности гибкая
При проведении ПКИ и РКИ есть существенные различия в методологии. В первую очередь, в ПКИ исследование осуществляется квалифицированными врачами, которые, однако, могут не иметь опыта в клинических исследованиях. При этом исследуется обширная группа пациентов, многие из которых не попали бы в РКИ в связи со строгими критериями включения. Таким образом, эти исследования могут помочь выяснить эффективность ЛП, например, у пожилых пациентов.
Несмотря на очевидные преимущества в получении реальных данных при проведении ПКИ, существуют определённые ограничения, которые необходимо разрешить.
Выбор центра исследования и протокола. Одним из серьёзных моментов при планировании ПКИ является выбор центра. Так, пациенты, которые регистрируются в одном центре, где планируется исследование, могут параллельно получать помощь и наблюдаться в других медицинских организациях, включая стационары, специализированные учреждения и амбулаторное звено. Это поднимает вопрос о необходимости качественного сбора всех соответствующих источников данных для получения полноценной информации о состоянии здоровья пациента [11]. При планировании ПКИ необходимым является участие гетерогенных центров, чтобы в исследовании приняли участие различные группы пациентов, и результаты были более представительны. Ещё одним важным вопросом до начала исследования является разработка протокола с учётом рутинной клинической практики, чтобы минимизировать вмешательства в ход рутинного лечебного процесса для наибольшей достоверности данных. Решить эту проблему можно путём привлечения врачей и персонала на ранней стадии разработки протокола [12].
Включение пациентов в исследование. Для включения пациента в классическое РКИ существуют строгие критерии отбора. Однако эти пациенты могут быть несопоставимы с пациентами из «реального мира», у которых присутствуют коморбидные патологии, тяжёлое течение заболевания, а также различные факторы риска [13]. С целью получения данных об эффекте исследуемого ЛП на более широкой популяции, в ПКИ используют более мягкие критерии включения. Количество критериев исключения желательно свести к минимуму. Однако, как и в других исследованиях, пациенты с абсолютными противопоказаниями не включаются в исследование. Традиционно возникают сложности с участием уязвимой группы населения. Однако принадлежность к таковой не должна являться критерием исключения. С учётом факта исследования уже зарегистрированного лекарственного препарата в ПКИ допускают участие таких пациентов с условием предоставления дополнительных мер защиты [14].
Уязвимые группы традиционно исключаются из предрегистрационных клинических исследований, поскольку они в меньшей степени способны защищать свои собственные интересы. Тем не менее, систематическое исключение из ПКИ препятствует нашему пониманию реальных преимуществ и вреда лекарственных препаратов, используемых этими группами.
Принадлежность к уязвимому населению сама по себе не должна быть критерием исключения из ПКИ, чтобы обеспечить равный доступ к знаниям, полученным в результате исследований. Предоставление этим группам возможности участвовать в ПКИ с помощью новых вмешательств даёт дополнительную выгоду от доступа к потенциально выгодному лечению. В настоящее время поощряется ответственное включение уязвимых лиц в ПКИ, если дополнительная защита обеспечивается там, где это считается необходимым, исходя из ожидаемых рисков, последствий и характеристик населения [14][15].
Рандомизация. Значительным отличием ПКИ от классического РКИ является и процедура рандомизации. Распределение пациентов в ПКИ осуществляется лечащим врачом (по возрастной группе, социальным признакам и так далее), а пациент обладает информацией о том, какой лекарственный препарат он принимает, или какое вмешательство осуществляется. Связанные с лечением процедуры исследования в ПКИ не должны менять рутинную медицинскую практику, поэтому решения о выборе дозировки ЛП, совместном вмешательстве и лечении нежелательных явлений оставляют на усмотрение лечащего врача [16, ][17]. Плацебо и другие методы ослепления пациентов и врачей для назначенной группы лечения, как правило, в ПКИ не используются, поскольку знание применяемого вмешательства, а также ожидания или изменения поведения, связанные с этими знаниями, являются частью эффекта лечения в реальной жизни. Кроме того, любые меры, принимаемые для содействия соблюдению режима лечения, должны отражать рутинную медицинскую практику [10][17].
Дизайн, где пациенты не имеют возможности выбрать предпочтительное вмешательство, часто рассматривается как барьер для участия в испытаниях [18], поэтому были предложены модификации процесса рандомизации. Одной из таких стал дизайн Зелена (предложен статистиком Высшей школы здравоохранения Гарварда Марвином Зеленом). В этой схеме пациенты рандомизируются либо в группу лечения, либо в контрольную группу прежде, чем дать информированное согласие. Поскольку группа, к которой относится данный пациент, известна, согласие может быть получено условно. Другим методом выступает кластерная рандомизация, предусматривающая назначение одинакового вмешательства определённой группе участников.
Информированное согласие. Одним из пунктов, ограничивающих участие пациента в ПКИ, является обширная и длительная процедура информированного согласия. При планировании таких исследований необходимо учитывать ограниченное количество визитов пациента в центр, что делает трудным проведение традиционной процедуры в условиях реальной медицинской практики. Также, в связи с ограниченным временем на приём пациента врачу представляется затруднительным длительное информирование пациента. Вышеперечисленное может привести к меньшему числу включённых пациентов в планируемое исследование, и менее репрезентативной выборке [19]. Учитывая это, был предложен ряд альтернатив для традиционного информированного согласия при проведении ПКИ, в которых сравниваются доказавшие свою действенность методы [20].
Интегрированное согласие. Модель предполагает обсуждение с пациентом предлагаемой терапии, существующих альтернатив, использование рандомизации, а также потенциальный вред и преимущества сравниваемого лечения [21]. Согласие пациента может быть получено устно или письменно. Врач документирует беседу и её результаты в электронную медицинскую карту (ЭМК).
Таргетированное согласие. Такая форма предлагается для сведения к минимуму вмешательства в реальную медицинскую практику при сравнении известных методов лечения [22]. Пациенту предлагается письменная форма согласия с указанием следующих данных: процедура и продолжительность исследования, инструкции по приёму ЛП, доступность исследуемого лечения для пациента вне рамок исследования, меры конфиденциальности, контактная информация и заявление об отказе от участия в исследовании в любое время без последствий. Разница с интегрированным согласием заключается в отсутствии информации о рандомизации [23].
Освещаемый подход. В этом случае используются общие уведомления, размещаемые на видных местах, информирующие пациентов о том, что они могут участвовать в сравнительных исследованиях эффективности со стандартными известными вмешательствами. Далее пациент сам может выразить желание об участии без явной процедуры письменного согласия, с учётом наличия минимальных рисков и информирования пациентов о возможном проведении в структуре здравоохранения интегрированных в практику исследований [24].
Отказ от согласия. В этом случае участники не проинформированы об исследовании и не принимают активного решения относительно участия. Теоретически такой вариант возможен для тех ПКИ, в которых все виды лечения, предлагаемые в исследовании, могут быть предложены за пределами испытания без специального информированного согласия, при этом лечение не связано с более чем минимальным дополнительным риском по сравнению с любой из альтернатив [25].
Отслеживание нежелательных лекарственных реакций. В ПКИ количество визитов пациента в центр и структурированный сбор данных сведены к минимуму, с целью минимизации вмешательства в рутинную медицинскую практику. Порядок отчётности о нежелательных явлениях (НЯ) и нежелательных лекарственных реакциях (НР) может оказать сильное вмешательство в этот процесс. В случае с прагматическими, порядок сбора отчётности может нарушаться, вследствие редкого посещения пациентов исследовательского центра. Также возникшая НР может привести к госпитализации пациента в другую клинику или обращению к другому специалисту, не связанному с исследовательским центром [26]. Поскольку в ПКИ задействованы врачи, которые могут не иметь опыта участия в исследованиях, у них может не быть опыта заполнения отчётности о НЯ, а также у них может быть недостаточная квалификация, чтобы связать возникновение НЯ с приёмом ЛП [27]. Решением в данном случае может стать применение ЭМК, однако данные системы должны быть валидированы для использования в исследовании [28]. Изменениям подвержен и традиционный подход к мониторингу клинического исследования. С учётом более широких критериев включения, число пациентов с рисками возрастает, однако частые визиты монитора нарушают рутинный медицинский процесс. Также мониторы могут не иметь полного доступа к ЭМК, что затруднит сбор данных. Вариантами решения данной проблемы может стать удалённый сбор данных, что избавит от необходимости регулярного посещения исследовательского центра [29]. Помимо этого, на этапе разработки протокола конкретного ПКИ требуется решить, какие данные и как часто будут необходимы для регулярной отчётности, а также каким образом обеспечить доступ к этим данным.
Преимущества и недостатки прагматических клинических исследований показаны в табл. 2.
Преимущества и недостатки прагматических клинических исследований