Миоклоническая эпилепсия

Миоклоническая эпилепсия — заболевание, основу которого оставляют миоклонические эпилептические пароксизмы. Эпизоды миоклонических судорог у больных сочетаются с генерализованными клонико-тоническими эпиприступами, абсансами. Сопутствующая неврологическая симптоматика зависит от формы эпилепсии. Диагностика включает сбор анамнеза, оценку неврологического и психического статуса, электроэнцефалографию, генеалогический анализ, биохимические исследования, нейровизуализацию. Лечение проводится антиконвульсантами, при резистентности — комбинацией противоэпилептических препаратов.
Общие сведения
Миоклонические судороги (миоклонии) представляют собой непроизвольные сокращения отдельной мышцы/мышечной группы. Соответственно, эпилепсия с преобладанием в клинической картине миоклоний получила название миоклоническая. Понятие «миоклоническая эпилепсия» (МЭ) включает ряд заболеваний, разнородных по этиопатогенезу, возрасту дебюта, особенностям симптоматики. В подавляющем большинстве случаев они характеризуется сочетанием миоклоний и генерализованных тонико-клонических судорожных приступов, имеют генетическую обусловленность. Встречаемость МЭ различна, некоторые нозологические формы являются настолько редкими, что в литературе по неврологии описано не более 100 клинических случаев.
Причины миоклонической эпилепсии
Обычно ведущим является генетический фактор. Чёткое аутосомно-доминантное наследование прослеживается при синдроме Драве, аутосомно-рецессивное — в отдельных случаях ранней миоклонической энцефалопатии. Некоторые заболевания имеют полигенное наследование. Локализация генетических дефектов установлена не для всех наследственных форм, исследования в этом направлении продолжаются. К генетически детерминированным патологиям относится и симптоматическая МЭ, возникающая вследствие дисметаболических процессов, обусловленных наличием дефектных генов. Образованию спонтанных мутаций в геноме способствуют:
Патогенез
Идиопатические варианты МЭ развиваются вследствие генетически обусловленной повышенной возбудимости церебральных нейронов, приводящей к эпилептогенной активности мозга. Симптоматическая миоклоническая эпилепсия формируется в результате обменных нарушений, накопления в нервных клетках патологических соединений (полисахаридных включений, прионных белков).
При болезни Лафоры, миоклонической энцефалопатии младенцев повышенная эпиактивность обусловлена дисметаболизмом нейронов в условиях разрастания глиальных элементов (при гибели нейронов, нарушении апоптоза астроцитов). Нейрональная гипервозбудимость вызывает возникновение патологической нервной импульсации, идущей к мышечным волокнам. Результатом являются отдельные мышечные сокращения (миоклонии), тонические, клонические судороги. Различная локализация миоклоний отражает локальное возбуждение разных зон мозговой коры. При диффузном распространении гипервозбуждения возникает клонико-тонический пароксизм с тотальным вовлечением мышечных групп.
Классификация
В основе группировки отдельных видов МЭ лежит этиологический принцип. Согласно Международной классификации эпилепсии 1989 года выделяют 3 основные группы:
Впоследствии были выявлены генетические аспекты возникновения криптогенных форм МЭ. Учитывая результаты исследований, Международное общество неврологов предложило относить ранее считавшиеся криптогенными виды МЭ к идиопатическим.
Симптомы миоклонической эпилепсии
Базовым симптомом выступают пароксизмы миоклоний, затрагивающие различные мышечные группы конечностей, реже — лица, еще реже — туловища. Миоклонии выглядят как мышечные подёргивания, при вовлечении мышц одной группы сокращения приводят к непроизвольным двигательным актам, напоминающим гиперкинезы. Миоклонический эпилептический пароксизм происходит при сохранённом сознании, может протекать с перемещением сокращений по различным мышцам. Миоклоническая эпилепсия характеризуется комбинацией миоклоний с клонико-тоническими приступами и/или абсансами. В зависимости от нозологической формы наблюдаются задержка психического развития, атаксия, пирамидная недостаточность, расстройства мышечного тонуса, зрительные нарушения.
ДМЭМ дебютирует в возрастном периоде от 6 месяцев до 3 лет. Приступы захватывают верхние конечности, лицо, шею, могут имитировать наклон головы, моргание, кивки головой. Заболевание редко сопровождается интеллектуальным снижением. Миоклоническая эпилепсия юношеского возраста (манифестация в возрасте 12-18 лет) отличается присоединением тонико-клонических эпизодов, отсутствием неврологического дефицита. Синдром Драве клинически проявляется на первом году жизни, сопровождается олигофренией, нарушениями поведения, пирамидным дефицитом. Семейная миоклония Унферрихта-Лундборга начинается в 5-16 лет, сочетается с тремором, атаксией, дизартрией, психическими расстройствами.
Миоклонически-астатические пароксизмы отличаются возникающей на фоне миоклоний потерей устойчивости. Пациенты описывают приступ как «удар под коленки», «подкашивание ног», вынуждающее становиться на колени, падать. Миоклонические абсансы представляют собой эпизоды кратковременного отключения сознания с миоклоническими сокращениями плечевого пояса, мышц конечностей, периорбитальной области. Заболевание возникает у детей 2-12 лет.
Миоклоническая эпилепсия симптоматического характера отличается прогрессированием симптоматики, выраженным когнитивным дефицитом, прочими неврологическими нарушениями, наличием проявлений основного заболевания, клинико-лабораторных признаков метаболических расстройств.
Осложнения
Клонико-тонические, астатические приступы осложняются травмированием пациента вследствие падения. Генерализованные судороги с утратой сознания опасны западением языка, перекрытием дыхательных путей и асфиксией. Аспирация слюны, рвотных масс приводит к последующему развитию пневмонии. Длительный миоклонический пароксизм, непрерывно следующие кластерные сокращения перерастают в миоклонический эпистатус. В эпилептическом статусе возможны серьёзные дыхательные расстройства, остановка сердца, развитие отёка головного мозга.
Диагностика
Миоклоническая симптоматика входит в клинику многих болезней, эпилептических синдромов. Диагноз «миоклоническая эпилепсия» устанавливается только при превалировании миоклонических приступов над другими клиническими проявлениями. Диагностика направлена на верификацию нозологической формы эпилепсии, при выявлении вторичного характера миоклоний — на поиск основной патологии. Основными диагностическими этапами являются:
Дифференциальная диагностика осуществляется с неэпилептическим миоклонусом, отличительной особенностью которого выступает фокальный характер миоклоний, отсутствие реакции на провокацию, нормальная ЭЭГ-картина. Дифференцировка МЭ необходима также с судорожным синдромом инфекционной этиологии, фебрильными судорогами, синдромом Леннокса-Гасто, мозжечковой миоклонической диссинергией Ханта.
Лечение миоклонической эпилепсии
Терапия базируется на антиконвульсантах. Подбор фармпрепарата и дозировки осуществляется индивидуально. Препаратами выбора выступают производные вальпроевой кислоты, обладающие противоэпилептическим эффектом в равной степени в отношении миоклонических, клонико-тонических, абсансных пароксизмов. В фармакорезистентных случаях показано комбинированное лечение вальпроатами, бензодиазепинами, этосуксимидом, барбитуратами, антиконвульсантами нового поколения (топираматом, леветирацетамом). Важным моментом является исключение провоцирующих приступы факторов: резких звуков, вспышек света, эмоциональных всплесков, физических перегрузок, перегреваний.
Прогноз и профилактика
Наиболее прогностически неблагоприятна ранняя миоклоническая энцефалопатия, смертность составляет половину случаев заболевания, остальные дети являются глубокими инвалидами. Миоклоническая эпилепсия при болезни Лафоры, Крейтцфельдта-Якоба плохо поддаётся противоэпилептической терапии, сопровождается прогрессирующим интеллектуальным распадом. ДМЭМ и ЮМЭ отличаются доброкачественным течением, редко приводят к когнитивному дефициту. Более 50% случаев ДМЭ заканчиваются спонтанным выздоровлением.
МЭ не имеет специфических мер профилактики. К мероприятиям, способным предупредить рождение больного ребёнка, относятся планирование беременности, ранняя постановка на учёт, исключение неблагоприятных воздействий на плод. Ведение беременности должно включать разъяснительные беседы с женщиной по поводу необходимости охранительного режима, тератогенной опасности лекарственных средств, пагубного воздействия на будущего ребёнка вредных привычек.
Миоклоническая эпилепсия
Миоклоническая эпилепсия — разновидность эпилептических приступов. Характеризуется более мягким течением. Впервые болезнь проявляется у младенцев или детей раннего возраста. Дебют миоклонической эпилепсии во взрослом возрасте нетипичен. Данная форма заболевания характеризуется вялым подергиванием мышц. Симптомы напоминают тики или гиперкинезы. В некоторых случаях возможно тяжелое течение. В международной классификации болезней МКБ-10 миоклоническая эпилепсия имеет код G40.3.
Пройти диагностику и лечение заболевания в Москве можно в Юсуповской больнице. Обследование проводят опытные неврологи и эпилептологи с использованием современного медицинского оборудования. Терапия соответствует европейским стандартам качества и безопасности.

Причины миоклонической эпилепсии
В настоящее время точные причины развития миоклонической эпилепсии не установлены. Однако, врачи выделяют несколько предрасполагающих факторов. Среди них:
Симптомы миоклонической эпилепсии
Клиническая картина миоклонической эпилепсии зависит от вида заболевания. Среди основных патологических симптомов выделяют:
Определение клинической симптоматики необходимо для подбора корректной терапии. Врачи Юсуповской больницы разрабатывают индивидуальный план лечения для каждого пациента.
Диагностика миоклонической эпилепсии
Миоклоническая эпилепсия требует проведения комплексной диагностики. Обследование включает в себя:
В Юсуповской больнице для диагностики миоклонической эпилепсии используется современное медицинское оборудование. Оно позволяет быстро и эффективно установить наличие заболевания. На основании полученных данных назначается корректная терапия.
Виды миоклонической эпилепсии
Миоклоническая эпилепсия делится на несколько видов. Рассмотрим основные из них.
Младенческая миоклоническая эпилепсия
Диагностируется в 30-40% случаев. Характеризуется симптомами по типу гиперкинезов или тиков. Слабая степень развития заболевания может протекать незаметно на протяжении многих лет. На фоне младенческой миоклонической эпилепсии не страдает интеллектуальное развитие. Заболевание может проявиться у ребенка с 2 месяцев до 3 лет.
Синдром Драве
Выявляется на первом году жизни. Симптоматически напоминает младенческую миоклоническую эпилепсию. Синдром Драве вызывает выраженные психические расстройства. Они проявляются олигофренией, умственной отсталостью. Без корректного лечения количество приступов увеличивается до нескольких раз в неделю.
Болезнь Унферрихт — Лундборга
Считается генетическим заболеванием. Характеризуется тяжелой неврологической симптоматикой. Болезнь Унферрихт — Лундборга также сопровождается психическими нарушениями. Первый приступ чаще всего проявляется в период полового созревания. Болезнь диагностируется в 10-20% случаев.
MERRF эпилепсия
Чаще диагностируется у взрослых. Миоклоническая эпилепсия юношеского возраста характеризуется тонико-клоническими пароксизмами. Болезнь не сопровождается неврологическими расстройствами, психическими нарушениями. Сознание на фоне приступа остается ясным.
Абсансы — разновидность эпилептического приступа. Считаются одним из симптомов миоклонической эпилепсии.
В зависимости от прогрессирования эпилепсии выделяют следующие формы болезни:
Лечение миоклонической эпилепсии
Для терапии миоклонической эпилепсии применяются медикаментозные средства. Врачи Юсуповской больницы применяют следующие группы препаратов:
Каждому пациенту разрабатывается индивидуальный план лечения. Он учитывает форму миоклонической эпилепсии, стадию развития, возраст больного и наличие сопутствующих заболеваний. Такой подход позволяет быстро и эффективно подобрать терапию. В Юсуповской больнице прием ведут опытные неврологи, эпилептологи, психиатры. Реабилитацию проводят квалифицированные массажисты, инструкторы ЛФК. Для того чтобы записаться на прием, необходимо позвонить по телефону или оставить заявку на официальном сайте больницы.
Абсанс

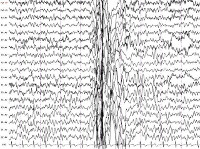
Абсанс — первично-генерализованный бессудорожный эпилептический припадок. Впервые описан в 1815 году Эскиролем под названием «малый эпилептический припадок» — petit mal (фр.). Его достоверный признак — специфическая ЭЭГ картина с билатерально-синхронной симметричной эпилептической активностью в виде комплексов пик-волна с частотой более 2,5 Гц (обычно 3,5–4 Гц) длительностью не менее 3 секунд. В классификации Международной противоэпилептической лиги 2017 года выделяют следующие виды абсансов:
типичный — характеризуется кратковременным нарушением сознания, при этом больной прерывает выполняемое действие, становится недоступен контакту, бледен, взгляд отрешен, направлен в одну точку. Припадок начинается и прекращается внезапно, длится около 10 секунд (варьируется от 1 до 44) и полностью амнезируется, пациенты говорят, что «задумались» и без затруднений возвращаются к занятию, которое прервал приступ.
миоклонический — сопровождается подергиванием мышц, как правило, лицевых или верхних конечностей
с миоклонией век — провоцируется закрыванием глаз, характеризуется подергиванием век и закатыванием глаз
Абсанс как разновидность приступа входит в клинические синдромы:
детская абсансная эпилепсия — самый распространенный эпилептический синдром в педиатрической практике. Изначально описан в медицинской литературе под названием «пикнолепсия», отмечен его наследственный характер. Дебютирует в возрасте от 4 до 10 лет, чаще всего в 5–7 лет. Неврологический статус в остальном нормальный, отставания в психическом развитии нет. В клинике синдрома — ежедневные многократные типичные абсансы. С возрастом может переходить в ювенильную абсансную эпилепсию.
ювенильная абсансная эпилепсия — дебютирует в возрасте от 10 до 20 лет, пик частоты в 10–12 лет. В клинике этого синдрома присутствуют и абсансы, и генерализованные тонико-клонические припадки (ГКТП), По сравнению с детской формой абсаснсной эпилепсии приступов в сутки меньше, нарушение сознания во время приступа менее выражено, но изменения на ЭЭГ сохраняются дольше. Эта форма также склонна к персистированию во взрослом возрасте.
абсансная эпилепсия с миоклонией — дебют в возрасте от 1 до 12 лет, чаще в 7 лет. Характеризуется ГТКП и абсансами с миоклонией, затрагивающей, как правило, мышцы верхних конечностей, длительность припадка 10–60с, частота — несколько раз в день.
миоклония век с абсансами — может быть симптоматической и идиопатической (синдром Дживонса). Для приступов характерна фотопровокация: возникают, например, при закрывании глаз.
Абсансную эпилепсию часто сопровождает психическая патология: по данным американских исследователей, у четверти детей выявляется когнитивный дефицит, у 43% — задержка речевого развития, у 61% есть психиатрический диагноз (наиболее распространенный — синдром дефицита внимания и гиперактивности, также встречаются депрессивные и тревожные расстройства). При этом риск психического расстройства тем выше, чем больше продолжительность болезни и частота припадков. Данные нейровизуализации позволяют предположить общий патогенез абсанса и коморбидных психических расстройств: так, характерные для абсанс-эпилепсии дисфункция таламуса и коры лобной доли, нарушения серотонинового обмена типичны также и для тревожных расстройств. Противоречивые данные получают при синдроме дефицита внимания и гиперактивности (СДВГ), коморбидном абсанс-эпилепсии: при самостоятельном СДВГ наблюдается снижение плотности коркового вещества, при СДВГ на фоне абсанс-эпилепсии она увеличена по сравнению с детьми, страдающими только абсанс-эпилепсией без СДВГ.
Стандарт диагностики абсанса — электроэнцефалогарафия. Другие методы применяются обычно в сочетании с ЭЭГ в исследовательских целях или для выбора тактики в случаях тяжелого течения и фармакорезистентности.
В частности, функциональная магнитно-резонансная томография позволяет подробнее изучить патогенез коморбидных изменений памяти, внимания, поведения, показывая функциональные изменения в нейронных сетях.
Транскраниальная магнитная стимуляция (ТМС) помогает определить степень возбудимости коры, причем исследования проводятся также на родственниках пациентов, позволяют выявить субклиническую гипервозбудимость и изучать наследование абсанс-эпилепсии. Также ТМС можно использовать для контроля эффективности фармакотерапии: при применении блокаторов кальциевых и натриевых каналов — повышение порога активации двигательного ответа, при лечении ГАМКергическими препаратами — повышение интракраниального ингибирования и снижение вероятности возбуждения.
Магнитоэнцефалография позволяет зарегистрировать высокочастотные осцилляции во время эпилептического приступа и локализовать их источник для возможного хирургического лечения. Метод представляет определенный научный интерес, поскольку до сих пор нет единого мнения, являются ли первоначальным источником патологического возбуждения при абсансе нейроны коры или таламуса; по данным, полученным с помощью МЭГ, источником является медиальная префронтальная кора.
Причины абсансных припадков — патология нейромедиаторной системы ГАМК и кальциевых каналов, что приводит к дисфункции таламо-кортикальных связей, очаги патологической активности обнаруживаются при этом как в таламусе, так и в коре (преимущественно лобных и теменных отделов).
Функцию ГАМК-А рецепторов по тоническому ингибированию облегчает и поддерживает активация ГАМК-В рецепторов: на животных показано, что введение их агонистов (например, баклофена) вызывает и усиливает припадки по типу абсанса. Таким действием обладают и некоторые антиконвульсанты, например, фенитоин и карбамазепин, поэтому при абсанс-эпилепсии они не рекомендуются. ГАМК-В рецепторы есть и в коре, и в таламусе, а также в гиппокампе (где участвуют в развитии атипичных абсансов).
Фазное ГАМК-ингибирование заключается в генерации ингибиторных постсинаптических потенциалов и реализуется за счет синаптических ГАМК-А рецепторов. Оно также является компонентом патогенеза абсанс-эпилепсии: в противоположность тоническому, фазное ингибирование в коре и таламокортикальных нейронах сенсорных ядер таламуса оказывается сниженным. У пациентов с абсанс-эпилепсией обнаруживаются многочисленные мутации ГАМК-А рецепторов, которые могут быть тому причиной.
Патогенез абсанс-эпилепсии в общих чертах выглядит так: сниженное фазное ГАМК-А ингибирование в коре может приводить к формированию осцилляций с частотой 5-9 Гц в первичной зоне возбуждения (которая могла бы стать мишенью для хирургического лечения). Возбуждение из коры распространяется на ретикулярные ядра таламуса, где также находятся ГАМК-ергические нейроны, которые в свою очередь вызывают генерацию тормозных постсинаптических потенциалов в таламокортикальных нейронах сенсорных ядер. Однако в пространстве вокруг таламокортикальных нейронов много свободной ГАМК, которую не утилизирует GAT-1. За счет этого, а также торможения со стороны ГАМК-В рецепторов, таламокортикальные нейроны подвергаются тоническому ингибированию. Они находятся в состоянии гиперполяризации, не способны возбуждаться в ответ на входящую сенсорную информацию и проводить ее к коре мозга, что клинически проявляется потерей сознания. Патологическая циркуляция возбуждения поддерживается за счет редких импульсов, доходящих до коры.
Терапия зависит от конкретного синдрома, в рамках которого встречается абсанс, однако должна удовлетворять общим принципам лечения эпилепсии. Монотерапия эффективна не при всех синдромах абсанс-эпилепсии, однако своевременному началу лечения и препарату, с которого начинается терапия, придается большое значение, поскольку от него может зависеть общее качество жизни ребенка в дальнейшем (успеваемость в школе, способность социализироваться). Индивидуальный подход и осторожное изменение дозировок важны потому, что детские эпилептические синдромы с возрастом могут проходить самостоятельно; лечение повышает вероятность и качество ремиссии, но невнимательная отмена препаратов может спровоцировать обострение.
При детской абсанс-эпилепсии рекомендованы этосуксимид, ламотриджин и препараты вальпроевой кислоты. Ни один из них не прошел испытания на I или II класс, но, особенно при простых абсансах, препаратом выбора считается этосуксимид; он также уменьшает проявления коморбидного СДВГ у таких детей. Из исследования, посвященного сравнению эффективности трех препаратов, в котором этосуксимид показывал наилучшие результаты, большинство пациентов вышли по причине плохой переносимости препарата. У 40% пациентов с возрастом к абсансам присоединяются ГТКП (в среднем через 5–10 лет после начала абсансов, в возрасте 8–15 лет; при этом факторами риска являются мужской пол, плохой ответ на первичную терапию, лечение единственным препаратов против абсансов), однако синдром читается доброкачественным и в 54–75% случаев удается достичь устойчивой ремиссии.
При юношеской абсанс-эпилепсии применяются препараты вальпроевой кислоты и ламотриджин (этосуксимид не рекомендуется, поскольку увеличивает частоту генерализованных тонико-клонических припадков, которые сочетаются с абсансами при этом синдроме). Также описано эффективное применение амантадина и кетогенной диеты в качестве мер второй линии. Расстройство чаще, чем детская абсанс-эпилепсия, сохраняется во взрослом возрасте; эффективность лечения и прогноз во многом зависят от локализации очага и распространения возбуждения, которые оцениваются дополнительными методами диагностики.
При абсанс-эпилепсии с миоклонией применяют комбинированную терапию: вальпроаты и этосуксимид или ламотриджин в комбинации с одним из этих препаратов. Отсутствия абсансов можно добиться со временем, но ГКТП остаются. Данная форма считается неблагоприятной, поскольку сопровождается снижением интеллекта.
Синдром Дживонса (миоклония век с абсансами) — фармакорезистентная форма эпилепсии, полное излечение практически невозможно. Основная мера по снижению количества приступов — избегание провоцирующих факторов (ношение темных очков). В качестве фармакотерапии применяют этосуксимид, препараты вальпроевой кислоты, бензодиазепины, левотирацетам и фенобарбитал. Для борьбы с фармакорезистентными формами также предлагают стимуляцию блуждающего нерва, глубокую стимуляцию мозга, транскраниальную магнитную стимуляцию.
Источники:
Fisher, R. S., Cross, J. H., French, J. A., Higurashi, N., Hirsch, E., Jansen, F. E., Lagae, L., Moshé, S. L., Peltola, J., Roulet Perez, E., Scheffer, I. E. and Zuberi, S. M. (2017), Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia, 58: 522–530. doi: 10.1111/epi.13670
Jeffrey R. Tenney and Tracy A. Glauser (2013) The Current State of Absence Epilepsy: Can We Have Your Attention?. Epilepsy Currents: May/June, Vol. 13, No. 3, pp. 135-140. doi: 10.5698/1535-7511-13.3.135
Abou-Khalil B. (2017) Other Pharmacological Therapies: Investigational Antiepileptic Drugs, Animal Models of Epilepsy, Hormonal Therapy, Immunotherapy. In: Koubeissi M., Azar N. (eds) Epilepsy Board Review. Springer, New York, NY. doi: 10.1007/978-1-4939-6774-2_19
Ravindra Arya et al. (2013) Vagus nerve stimulation for medically refractory absence epilepsy. Seizure, Volume 22, Issue 4, May 2013, Pp. 267-270 doi: 10.1016/j.seizure.2013.01.008
Neuroscience Letters, Volume 566, 30 April 2014, Pages 21-26; Using ictal high-frequency oscillations (80–500 Hz) to localize seizure onset zones in childhood absence epilepsy: A MEG study; Miao et al.
Epilepsia. 2008 Nov;49(11):1838-46. doi: 10.1111/j.1528-1167.2008.01680.x. Epub 2008 Jun 13. Childhood absence epilepsy: behavioral, cognitive, and linguistic comorbidities; Caplan R1, Siddarth P, Stahl L, Lanphier E, Vona P, Gurbani S, Koh S, Sankar R, Shields WD.
Journal of the Neurological Sciences, Volume 339, Issues 1–2, 15 April 2014, Pages 189-195; Altered intrinsic functional connectivity of the salience network in childhood absence epilepsy; Cheng Luo et al.